近年来,多肽药物凭借其高特异性、低毒性以及能够靶向传统"不可成药"靶点的优势,在创新药研发领域备受瞩目。尤其是环肽(Cyclic Peptides),由于其特殊的环状结构,具备了更好的体内稳定性和穿透细胞膜的潜力。
然而,传统的环肽筛选往往依赖于耗时耗力的物理库构建与实验试错。随着计算生物学的飞速发展,AI算法与分子对接技术的结合正在重塑这一过程。今天,我们将以**科晶生物推出的算法设计靶向高亲和环肽技术服务**为案例,深度解析如何利用前沿算法,高效完成从头设计到精准筛选的全流程。
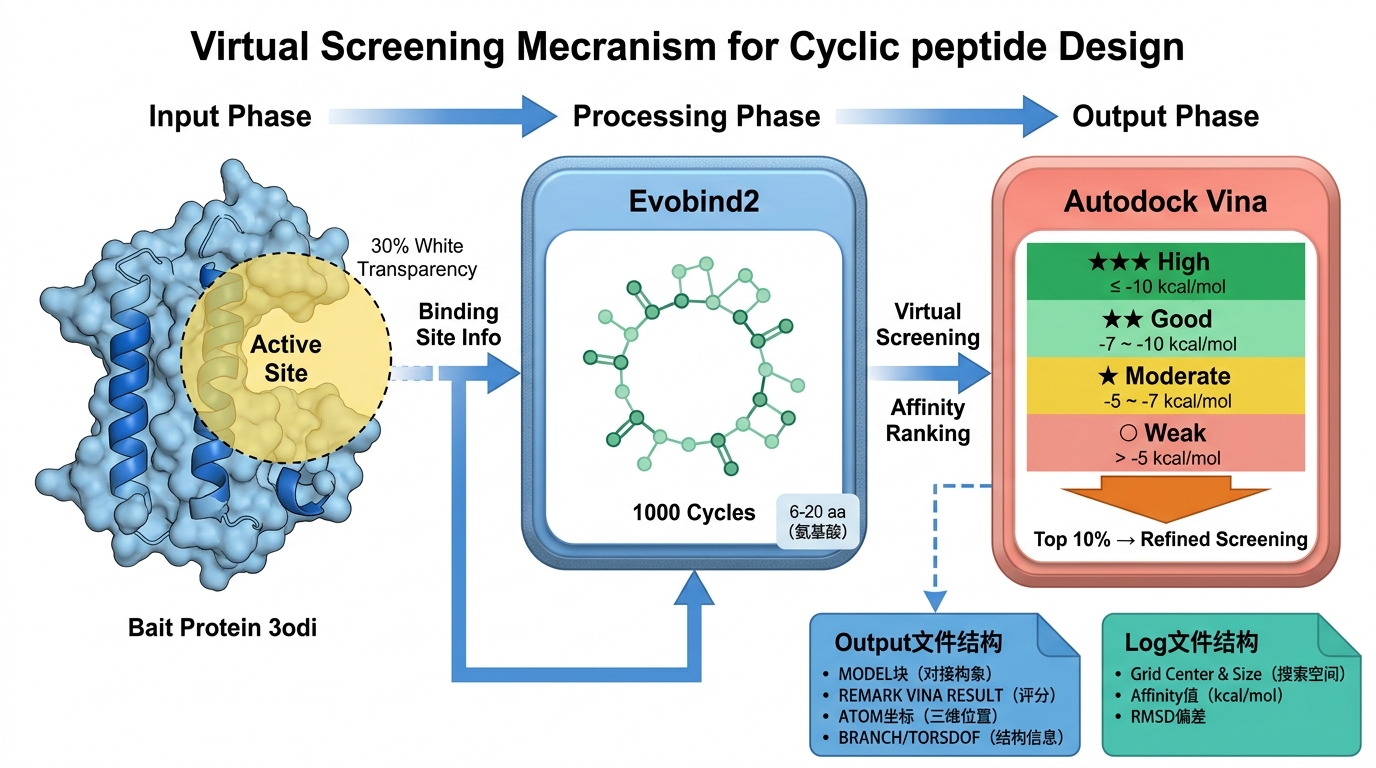
科晶生物环肽设计技术流程纵览
第一步:精准锚定靶标结合位点
药物设计的核心在于"对症下药"。在科晶生物的技术实践中(以诱饵蛋白3odi为例),第一步并非盲目生成序列,而是基于靶点蛋白晶体结构,精准定位其与底物结合的活性区域。该区域将被设定为未结合位点区域,作为后续环肽设计的"定制口袋"。这种带有明确靶向性的空间限定,为后续的高亲和力结合奠定了基础。
第二步:Evobind2 赋能,完成环肽"从头设计"
确定口袋后,面临的挑战是如何生成适配该完整三维空间的分子。在这一阶段,引入了前沿的 Evobind2 模型。
根据设定的参数,Evobind2 能够自动设计出长度在6-20个氨基酸之间的环肽序列。相对于传统方法,Evobind2 的核心优势在于其高效、精准与自动化。在短时间内,由该算法构建出包含1000个高潜力环肽候选物的虚拟库。这不仅极大地提升了早期药物发现阶段的筛选速度,更从源头上优化了环肽的结合亲和力,让"大海捞针"变成了"按图索骥"。
第三步:基于 AutoDock Vina 的高通量与精细化筛选
生成了1000个候选环肽后,真正考验技术功底的是如何把"最好用"的分子挑出来。这时候,经典的分子对接软件 AutoDock Vina 成为了核心过滤网。
AutoDock Vina 通过改进的评分函数(Scoring function),对环肽与蛋白质的三维相互作用进行物理化学层面的打分。科晶生物的筛选策略体现了极强的工程化思维:
- 初筛识别: 快速遍历1000个环肽,通过计算结合自由能(Max Affinity),初步识别出稳定性好、亲和力强的高潜力互作。
- 精细化对接验证: 严格提取初筛库中 Max Affinity 排名前10% 的"优等生",进入计算量更大、精度更高的精细化筛选。结合能量越低(负值越大),对接预测越准确,最终锁定与靶蛋白高度互作的候选分子。
技术延伸:我们如何定义"好"的环肽?
在获取了对接结果后,数据的解读是判断药物潜力的关键环节。在实际项目输出中,通过多维度的指标来衡量环肽的优劣:
- 亲和力梯度(Affinity): 这是最直观的指标。通常,结合自由能在 -10 kcal/mol 或更低,代表着非常好的结合(High affinity);在 -7 到 -10 kcal/mol 之间属于良好结合;-5 到 -7 kcal/mol 为中等结合;大于 -5 kcal/mol 则是弱结合。
- 能量拆解(INTER + INTRA): 结合不只要"紧",还要"稳"。通过考量分子间作用力(INTER,即环肽与靶蛋白的结合力)与分子内能量(INTRA,即环肽自身的构象应变能),确保筛选出的分子在维持结合强度的同时,自身结构也没有过度扭曲变形。
- 构象一致性(RMSD): 报告输出通常选取结合能量最低的最优构象(Mode 1),并计算其他构象偏离最优构象的均方根偏差(RMSD)。偏离度越小,说明该对接模式在热力学系统下越稳定可靠。
结语:从"实验发现"到"理性设计"
从蛋白口袋的精准定位,到 Evobind2 的大批量生成,再到 AutoDock Vina 的严苛筛选。我们可以清晰地看到,多肽药物研发正在由传统的"试错式实验"向计算机辅助的"理性设计"快速转型。
类似科晶生物所提供的此类高亲和环肽算法设计服务,本质上是在为生物医药企业和科研工作者提供一套"外脑引擎"。它不仅大幅压缩了新药研发早期的资金和时间成本,更通过高并发的算力与精准的AI模型,为攻克疑难疾病靶点提供了源源不断的优质候选分子。在AI大模型的浪潮下,我们有理由相信,下一个重磅多肽药物,或许就诞生在代码与算力的交汇处。
参考文献:
1 Li Q, Vlachos E.N., Bryant P. Design of linear and cyclic peptide binders of different lengths from protein sequence information. bioRxiv. 2024. p. 2024.06.20.599739.
2 Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455-461.
3Seeliger, D., & de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and AutoDock/Vina. Journal of Computer-Aided Molecular Design, 24(5), 417-422.
4O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. Journal of Cheminformatics, 3(1), 33.