目录
1,数据输入及处理
①载入包和数据
官方学习:focuslyj/CellChat - 码云 - 开源中国 (gitee.com)
这里提供的应该是counts data数据
rm(list=ls())
library(CellChat)
library(patchwork)
library(ggplot2)
library(Seurat)
library(ggalluvial)#绘制桑基图
library(expm)
library(sna)
library(NMF)
options(stringsAsFactors = FALSE)##输入数据不自动转换成因子(防止数据格式错误)
load("data_humanSkin.Rdata")#数据加载:这里是count data数据②CellChat输入数据准备
#需标准化的基因表达量矩阵和细胞分组信息文件
#不同输入格式处理方式不同
data.input = data_humanSkin$data#需标准化的基因表达量矩阵和细胞分组信息文件
meta = data_humanSkin$meta
data.input[1:6,1:3]#表达count
head(meta);table(meta$condition) #含normal(NL)和diseases(LS)
cell.use = rownames(meta)[meta$condition == 'LS'] #提取LS的细胞名称
data.input = data.input[, cell.use]#提取LS表达矩阵
meta = meta[cell.use, ]#提取LS细胞信息
identical(rownames(meta),colnames(data.input)) #检查矩阵列名和分组文件行名是否一致
unique(meta$labels) #检查细胞亚群标签类型③构建CellChat对象
#接下来构建CellChat对象
cellchat <- createCellChat(object = data.input, #支持normalized表达矩阵,Seurat对象,和SingleCellExperiment对象
meta = meta, #meta文件
group.by = 'labels') #meta中的细胞分类列
#cellchat <- addMeta(cellchat, meta = meta)#创建CellChat对象未 cellmeta信息时添加信息
cellchat <- setIdent(cellchat, ident.use = 'labels') #将label设置为显示的默认顺序
levels(cellchat@idents) #查看celltype和factor顺序
table(cellchat@idents) #每个celltype中的细胞数
#设置配受体数据库(CellChatDB):
CellChatDB <- CellChatDB.human #(CellChatDB.human) (CellChatDB.mouse)
showDatabaseCategory(CellChatDB) #查看描述该数据库组成的饼状图
dplyr::glimpse(CellChatDB$interaction) #查看数据库结构
#直接使用CellChatDB全库进行细胞通讯分析:
##CellChatDB.use <- CellChatDB # simply use the default CellChatDB
#选择数据库中特定子集进行细胞通讯分析:
CellChatDB.use <- subsetDB(CellChatDB,
search = 'Secreted Signaling') #可选择Secreted Signaling、ECM-Receptor或Cell-Cell Contact
cellchat@DB <- CellChatDB.use#将数据库添加到CellChat对象中(DB)④数据预处理
#数据预处理;信号基因的表达矩阵子集化,节省计算成本
cellchat <- subsetData(cellchat) #必选的step,取上一步CellChatDB.use中信号基因的表达矩阵子集,赋值到cellchat@data.Signaling
#future::plan('multiprocess', workers = 4) # do parallel (可以不用选择平行计算)
#鉴定与每个细胞亚群相关的过表达信号基因:基于表达该基因的细胞比例、差异倍数和p值判定。
cellchat <- identifyOverExpressedGenes(cellchat,
only.pos = TRUE, #仅返回positive markers
thresh.pc = 0, #细胞比例阈值
thresh.fc = 0, #差异倍数
thresh.p = 0.05) #P-Value
#计算结果赋值到cellchat@var.features:
head(cellchat@var.features$features) #过表达信号基因名
head(cellchat@var.features$features.info) #差异计算结果表
#识别过表达基因配体-受体互作:
cellchat <- identifyOverExpressedInteractions(cellchat)
head(cellchat@LR$LRsig) #计算结果赋值位置
#将基因表达数据映射到PPI网络(可跳过):
cellchat <- projectData(cellchat, PPI.human) #返回结果:cellchat@data.project2,细胞通讯预测
①计算细胞通讯概率
#细胞通讯预测##############################################
cellchat <- computeCommunProb(cellchat, raw.use = TRUE) #返回结果:cellchat@options$parameter
##默认使用原始表达数据(cellchat@data.Signaling),若想使用上一步PPI矫正数据,设置raw.use = TALSE
cellchat <- filterCommunication(cellchat, min.cells = 10) #细胞通讯过滤(设置每个亚群中进行细胞间通讯所需的最小细胞数)②提取配受体对细胞通讯结果表
#提取配受体对细胞通讯结果表:
df.net <- subsetCommunication(cellchat, slot.name = 'net')
head(df.net) #得到配受体对细胞通讯结果表
#或访问其它感兴趣/特定的细胞通讯结果:
df.net1 <- subsetCommunication(cellchat,
sources.use = c('LC'),
targets.use = c('FBN1+ FIB')) #访问特定细胞对子集
head(df.net1)
df.net2 <- subsetCommunication(cellchat, signaling = c('CD40')) #访问特定信号通路子集
head(df.net2)③提取信号通路水平的细胞通讯表
提取配受体对细胞通讯结果表:subsetCommunication函数
提取信号通路水平的细胞通讯表:computeCommunProbPathway函数
#提取信号通路水平的细胞通讯表:
cellchat <- computeCommunProbPathway(cellchat) #计算信号通路水平上的通讯概率
df.netp <- subsetCommunication(cellchat, slot.name = 'netP') #得到信号通路水平细胞通讯表
head(df.netp)④细胞互作关系可视化
1)细胞亚群间配受体数目网络图
cellchat <- aggregateNet(cellchat)#计算细胞对间通讯的数量和概率强度
#不同细胞亚群间的互作数量与概率/强度可视化:
groupSize <- as.numeric(table(cellchat@idents))##细胞亚群间配受体数目网络图:
par(mfrow = c(1,1), xpd = TRUE)
netVisual_circle(cellchat@net$count,
vertex.weight = groupSize,
weight.scale = T,
label.edge = F,
title.name = 'Number of interactions')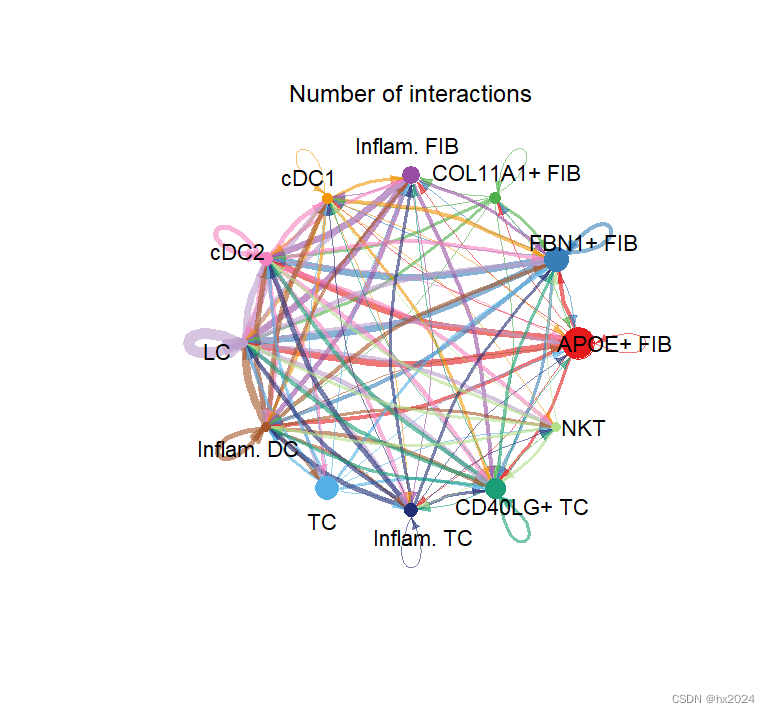
细胞通讯| 02.CellChat基础分析教程_哔哩哔哩_bilibili
2)细胞亚群间配受体概率/强度网络图
##细胞亚群间配受体概率/强度网络图:
par(mfrow = c(1,1), xpd = TRUE)
netVisual_circle(cellchat@net$weight,
vertex.weight = groupSize,
weight.scale = T,
label.edge= F,
title.name = 'Interaction weights/strength')
3)每个细胞亚群的配受体通讯概率进行单独展示
这里需要注意R绘图画板范围,可以将前面的绘图进行保存和devoff后再进行作图
#检查单个细胞亚群的互作信号强度;每个细胞进行单独展示##
mat <- cellchat@net$weight
par(mfrow = c(3,4), xpd = TRUE)
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(mat2, vertex.weight = groupSize, weight.scale = T, edge.weight.max = max(mat), title.name = rownames(mat)[i])
} 4)数量和强度弦图合并
4)数量和强度弦图合并
#数量和强度弦图合并
par(mfrow = c(1,2), xpd = TRUE)
netVisual_circle(cellchat@net$count,
vertex.weight = groupSize,
weight.scale = T,
label.edge = F,
title.name = 'Number of interactions')
netVisual_circle(cellchat@net$weight,
vertex.weight = groupSize,
weight.scale = T,
label.edge= F,
title.name = 'Interaction weights/strength')
#保存cellchat对象:
save(cellchat, groupSize, file = c('humanSkin_CellChat.Rdata'))3,信号通路水平的细胞通讯分析
cellchat@netP$pathways##信号通路查看
pathways.show <- c('GALECTIN')##以'GALECTIN'信号通路展示为例
#层级图(Hierarchy plot)绘制
levels(cellchat@idents)#查看细胞亚群及factor顺序:
#选择其中感兴趣的细胞亚群:
vertex.receiver = c(3,8,9,10)#为画图第一列的source列
par(mfrow = c(1,1))
netVisual_aggregate(cellchat,
layout = c('hierarchy'), #"circle", "hierarchy", "chord"
signaling = pathways.show,
vertex.receiver = vertex.receiver)#选择展示的通路
别的图
par(mfrow = c(1,1))#展示网络图
netVisual_aggregate(cellchat,
layout = c('circle'),
signaling = pathways.show)
par(mfrow=c(1,1))#展示弦图
netVisual_aggregate(cellchat,
layout = c('chord'),
signaling = pathways.show)
par(mfrow=c(1,1))#展示热图
netVisual_heatmap(cellchat,
signaling = pathways.show,
color.heatmap = c("white", "#b2182b"))
4,信号通路相关配受体对水平的细胞通讯分析
netAnalysis_contribution(cellchat,
signaling = pathways.show) #配受体对贡献条形图
pairLR.CXCL <- extractEnrichedLR(cellchat, #提取细胞对
signaling = pathways.show,
geneLR.return = FALSE)
LR.show <- pairLR.CXCL[1,] #以贡献度top1的配受体对为例
pairLR.CXCL; LR.show
别的图
netVisual_individual(cellchat,#Hierarchy plot:
layout = c('hierarchy'),
signaling = pathways.show, #目标信号通路
pairLR.use = LR.show, #目标配受体对
vertex.receiver = vertex.receiver) #感兴趣的细胞亚群
#Circle plot:
netVisual_individual(cellchat,
layout = c('circle'),
signaling = pathways.show,
pairLR.use = LR.show)
#Chord diagram:
netVisual_individual(cellchat,
layout = c('chord'),
signaling = pathways.show,
pairLR.use = LR.show)
5,多个配受体对/信号通路水平介导的细胞通讯可视化
①指定信号通路
多个配受体对/信号通路水平介导的细胞通讯可视化######
levels(cellchat@idents)#指定信号通路
netVisual_bubble(cellchat,
sources.use = 4,
targets.use = c(5:11),
signaling = c("CCL","CXCL"), #指定CCL和CXCL两个信号通路
remove.isolate = FALSE)
#指定配受体对:
pairLR.use <- extractEnrichedLR(cellchat,
signaling = c("CCL","CXCL","FGF")) #确定在目标信号通路中有重要作用的配受体对
pairLR.use
netVisual_bubble(cellchat,
sources.use = 4,
targets.use = c(5:11),
pairLR.use = pairLR.use,#指定的受体对
remove.isolate = TRUE)
②参与目标信号通路的基因在各细胞亚群的表达分布展示
#参与目标信号通路的基因在各细胞亚群的表达分布展示:
plotGeneExpression(cellchat, signaling = 'GALECTIN',
type = 'violin') #小提琴图
③气泡图
plotGeneExpression(cellchat,
signaling = 'GALECTIN', type = 'dot', color.use = c("white", "#b2182b")) #气泡图