书接上文,我们昨天讨论了遗传的哈代温伯格比例:
【基于R语言群体遗传学】-1-哈代温伯格基因型比例-CSDN博客
接下来,如果我们能够模拟一个过程并观察模拟结果与我们预期的是否相符,这通常有助于指导我们对这个过程的直观感觉。让我们来模拟哈代-温伯格基因型比例。首先,我们将创建一个等位基因列表,用于组成一个种群。
R
allele <- c(rep("A",2), rep("a",8))
allele
接下来,我们创建一个矩阵来记录种群中个体的等位基因。我们从一个变量popsize开始,最初设置为100,稍后我们可以轻松地更改它以模拟不同的人口规模。 然后,我们使用matrix()函数创建一个100行(由nrow = popsize定义)和两列(ncol = 2,二倍体)的矩阵,并将其保存为pop。
R
popsize <- 100
pop <- matrix(nrow=popsize, ncol=2)每一行对应一个个体 ,两列代表每个位点的两个副本,我们将在其中记录该个体的等位基因。 现在,我们将使用R的内置sample函数随机选择等位基因来构成我们的个体基因型。
R
for(i in 1:popsize){
pop[i,1] <- sample(allele,1)
pop[i,2] <- sample(allele,1)
}
pop我们得到一个不是特别规范的100个个体的基因型随机数据模拟,里面有杂合及纯合。

我们通过循环得到A和a的计数,并计算A的frequency和a的frequency
R
#A frequency
Acount <- 0
for(i in 1:popsize){
if(pop[i,1]=="A"){
Acount <- Acount+1
}
if(pop[i,2]=="A"){
Acount <- Acount+1
}
}
AFreq <- Acount/(popsize*2)我们计算得到A的frequency为0.375,大家可能有所不同,但是都会在这附近。现在,我们将通过再次遍历矩阵并计算杂合子出现的频率以及A/A纯合子出现的频率来计算基因型频率。
R
Hcount <- 0
AAcount <- 0
for(i in 1:popsize){
if(pop[i,1]=="A"){
if(pop[i,2]=="a"){
Hcount <- Hcount+1
}else{
AAcount <- AAcount+1
}
}
if(pop[i,1]=="a"){
if(pop[i,2]=="A"){
Hcount <- Hcount+1
}
}
}
HetFreq <- Hcount/(popsize)
AAFreq <- AAcount/(popsize)
print(c(AFreq, HetFreq, AAFreq))
现在,让我们将这些模拟的频率与所有三种基因型(A/A、A/a和a/a)的p^2、2p(1-p)和(1-p)^2预测一起绘制出来。以下命令将使用plot()函数绘制这些点。并添加曲线进行拟合:
R
# 绘制第一个图形
plot(AFreq, HetFreq, xlab="allele frequency", ylab="",
ylim=c(0, 1), xlim=c(0, 1), col="blue")
# 在同一个图形上添加第二个图形,不覆盖第一个图形
par(new=TRUE)
plot(AFreq, AAFreq, xlab="", ylab="", ylim=c(0, 1), xlim=c(0, 1), col="green")
# 在同一个图形上添加第三个图形,不覆盖前两个图形
par(new=TRUE)
plot(AFreq, 1-AAFreq-HetFreq, xlab="",
ylab="genotype frequency", ylim=c(0, 1), xlim=c(0, 1),
col="red")
# 设置点的样式,使其变大并实心
points(AFreq, HetFreq, pch=16, cex=2, col="blue") # 蓝色实心点
points(AFreq, AAFreq, pch=16, cex=2, col="green") # 绿色实心点
points(AFreq, 1-AAFreq-HetFreq, pch=16, cex=2, col="red") # 红色实心点
# 在同一个图形上添加三条曲线
curve(2*x*(1-x), 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkblue")
curve(x**2, 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkgreen")
curve((1-x)**2, 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkred")
# 添加文本标签
text(0.5, 0.7, "Aa", col = "blue")
text(0.9, 0.7, "AA", col = "green")
text(0.1, 0.7, "aa", col = "red")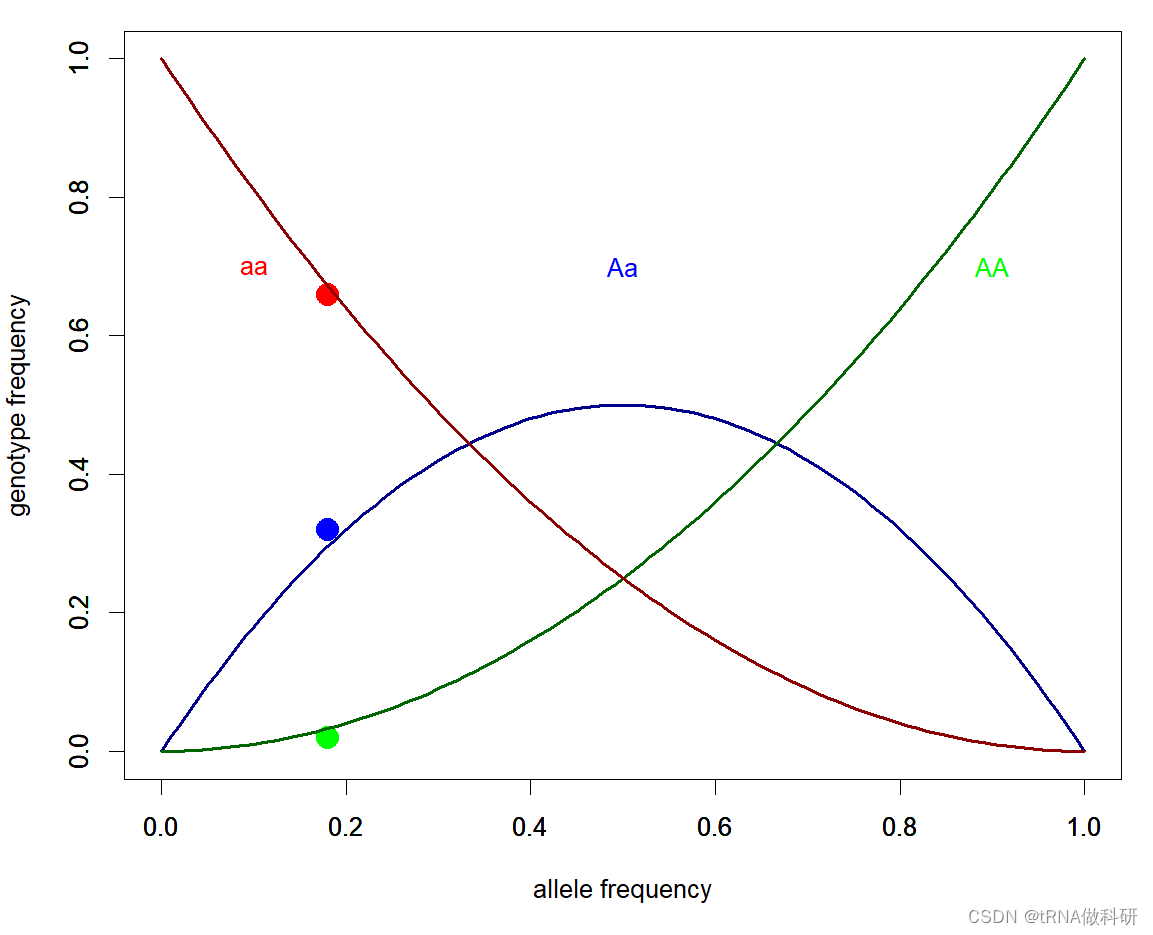
我们通常对许多点的分布感兴趣。让我们修改代码,让它运行一些重复实验并同时绘制它们。每次我们运行模拟时,我们都会抽取一个新的等位基因频率,因此我们运行的重复实验次数越多,我们将看到的等位基因频率范围就越广。
R
AFreq <- numeric()
HetFreq <- numeric()
AAFreq <- numeric()
replicates <- 10
for (j in 1:replicates) {
# Randomly sample alleles for each individual
pop[, 1:2] <- t(replicate(popsize, sample(allele, 2)))
# Count the number of 'A' alleles
Acount <- sum(pop == "A")
# Calculate the frequency of 'A' alleles
AFreq[j] <- Acount / (popsize * 2)
# Count the number of heterozygotes and AA homozygotes
genotypes <- apply(pop, 1, function(ind) {
if (all(ind == "A")) {
"AA"
} else if (any(ind == "A")) {
"Het"
} else {
"aa"
}
})
# Calculate the frequencies of heterozygotes and AA homozygotes
HetFreq[j] <- sum(genotypes == "Het") / popsize
AAFreq[j] <- sum(genotypes == "AA") / popsize
}我们如此得到一系列点,然后进行可视化:
R
plot(AFreq, HetFreq, xlab="allele frequency",ylab = "",
ylim=c(0, 1), xlim=c(0, 1), col="blue")
par(new=TRUE)
plot(AFreq, AAFreq, xlab="", ylab = "", ylim=c(0, 1), xlim=c(0, 1), col="green")
par(new=TRUE)
plot(AFreq, 1-AAFreq-HetFreq,xlab = "",
ylab="genotype frequency", ylim=c(0, 1), xlim=c(0, 1),
col="red")
# 绘制第一个图形
plot(AFreq, HetFreq, xlab="allele frequency", ylab="",
ylim=c(0, 1), xlim=c(0, 1), col="blue")
# 在同一个图形上添加第二个图形,不覆盖第一个图形
par(new=TRUE)
plot(AFreq, AAFreq, xlab="", ylab="", ylim=c(0, 1), xlim=c(0, 1), col="green")
# 在同一个图形上添加第三个图形,不覆盖前两个图形
par(new=TRUE)
plot(AFreq, 1-AAFreq-HetFreq, xlab="",
ylab="genotype frequency", ylim=c(0, 1), xlim=c(0, 1),
col="red")
# 在同一个图形上添加三条曲线
curve(2*x*(1-x), 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkblue")
curve(x**2, 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkgreen")
curve((1-x)**2, 0, 1, add=TRUE, ylab=NULL, lwd=2, ylim=c(0, 1), col="darkred")
# 添加文本标签
text(0.5, 0.7, "Aa", col = "blue")
text(0.9, 0.7, "AA", col = "green")
text(0.1, 0.7, "aa", col = "red")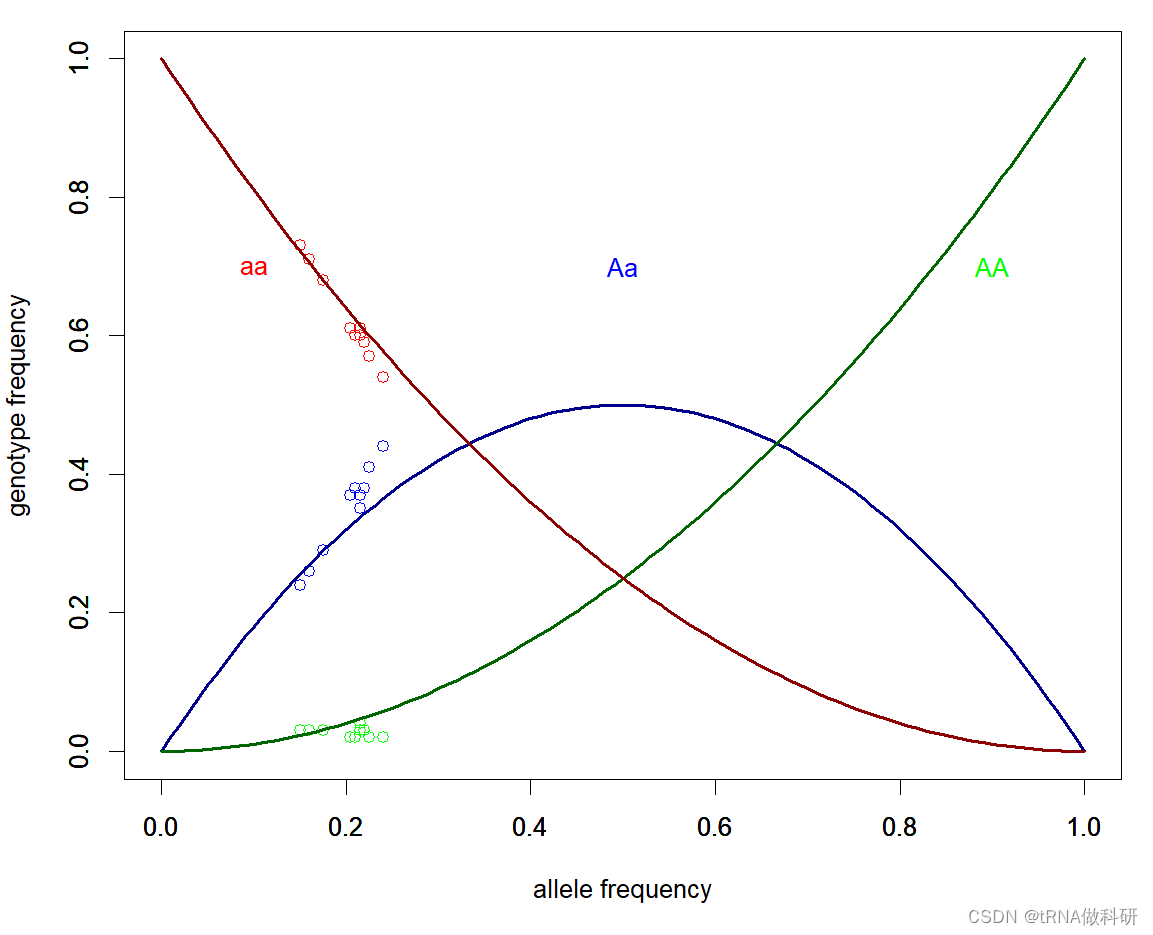
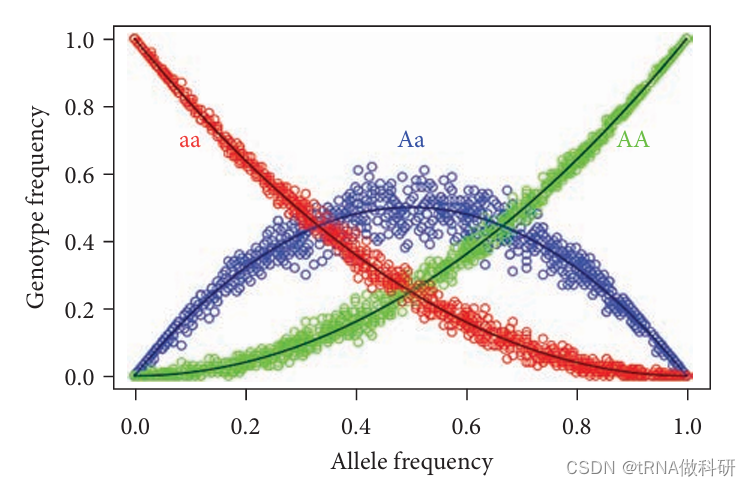
目前为止,这些模拟是通过从原始的"等位基因"列表(总共十个中有两个"A")中以p = 0.2为中心抽样等位基因频率来运行的。然而,我们可以修改代码,使其在整个可能的等位基因频率范围内运行。我们使用runif
R
p <- runif(1)
for(i in 1:popsize){ #Randomly sample alleles
pop[i,1] <- sample(allele,1)
pop[i,2] <- sample(allele,1)
}
for(i in 1:popsize){
if(runif(1)<p){
pop[i,1] <- "A"
}else{
pop[i,1] <- "a"
}
if(runif(1)<p){
pop[i,2] <- "A"
}else{
pop[i,2] <- "a"
}
}
当然 大家可以通过改变种群大小去看看差异
下一章将讲解R语言计算等位基因频率相关的内容,欢迎大家点赞收藏。