wget+网址用于直接从网上下载某个文件到服务器,当然也可以直接从网上先把东西下到本地然后用filezilla这个软件来传输到服务器上。
当遇到不会的命令时候,可以使用man "不会的命令"来查看这个命令的详细信息。比如我想要看看ls这个命令的详细用法,可以写man ls,结果如下,按q退出
ls用于查看当前目录下有什么
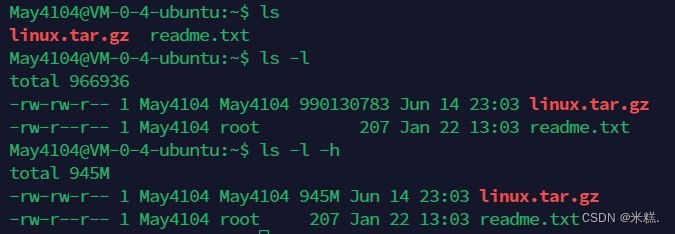
其中ls是命令,-l也好,-h也好,都是命令的修饰,如果只写一个ls就只显示当前目录下的文件名,加上-l表示以列表的形式查看当前文件的详细信息,再加上-h表示简洁的显示出文件的大小,可以看到这里的文件是945M。
在Linux系统中,ls命令用于列出目录的内容。ls -alF命令是ls命令的一个变体,其中包含了几个选项:
- -a:表示显示所有文件,包括以点(.)开头的隐藏文件。
- -l:表示以长列表格式显示文件信息,包括文件权限、所有者、大小、最后修改时间等。
- F:这个选项会在文件名后添加一个字符以指示文件类型。例如,/ 表示目录,* 表示可执行文件。
综合来看,ls -alF命令会以长列表格式显示当前目录下的所有文件(包括隐藏文件),并为每个文件名添加一个指示文件类型的字符。这个命令还被Linux系统起了一个别名,叫做ll,我们可以使用alias命令来查看系统都给哪些命令起了别名,如图
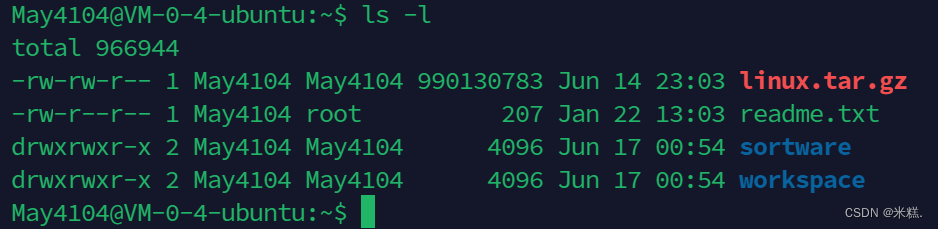
如何找到一个文件的地址?实际上文件的地址=文件所在的文件夹地址+本文件的文件名,而使用pwd我们可以得到当前文件所在的路径,比如我现在有一个文件叫linux.tar.gz,我运行pwd将得到这个文件所在的文件夹,
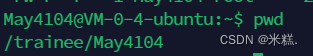
则显然该文件的绝对路径就是/trainee/May4104/linux.tar.gz
有了文件的路径,就可以配合ls命令来显示一个指定文件的信息了。
实际上我们可以写ls -lh,与上面的ls -l -h是等效的。
rm用于删除文件,但不能删除文件夹,删除文件夹使用的命令是rm -r +要删除的文件名,那么如何来看要删除的是文件还是文件夹呢?答案是使用ls -l以列表形式展示当前目录的所有内容(实际上直接ll就行),第一列以'-'开头的就是文件,以d开头的就是文件夹。
有时候我们使用rm -r来删除文件夹的时候,系统会一直问说是否要删除这个文件,我们要回答yes或者no,如果要删的文件夹里面有100个文件,我们就要打100次yes或者no,此时可以使用rm -r -f 来强制删除。
小键盘的上下键用于翻阅上下命令,这个技巧非常有用,可以避免我们重复的输入代码。
先选中某个命令,然后ctrl+shift+c/v表示复制或者粘贴
ctrl+c表示终止当前正在执行的命令
清空屏幕可以使用命令clear或者直接ctrl+l
cd /工作路径 用于切换工作目录,如果不指定具体工作路径只是有一个/,就是切换工作路径到根目录,也就是最大的目录,如果连/都没有,那就是切换到家目录。在Linux中有很多特殊路径有特殊的符号来表示,比如/表示跟目录,~表示家目录,一个点表示当前目录,两个点表示上一级目录。当然如果是上上级目录就是两组上级目录,也就是../..
建文件夹的命令是mkdir
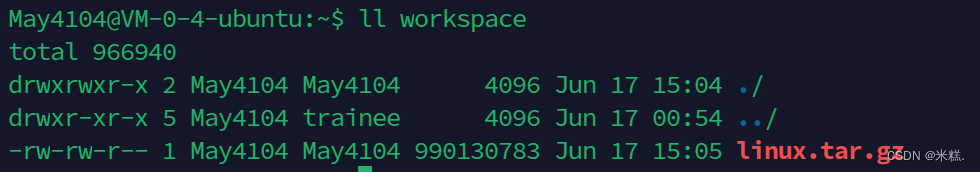
拷贝的命令是cp,比如我想要把linux.tar.gz这个文件拷贝到文件夹workspace中去,就可以写cp ,ll workspace就可以看见workspace下就变成了这样子。
cp命令还有一种用法,比如cp linux.tar.gz 123 其中123其实是不存在的一个文件夹,但是仍然能拷贝,此时这个命令的意思就是把 linux.tar.gz拷贝一份并命名为123
移动文件的命令是mv,比如要把linux.tar.gz移动到morkspace中去就写mv linux.tar.gz morkspace 注意是先写要移动的文件,再写把这个文件移动到哪里去。
mv也有另一种用法,比如cp linux.tar.gz 123 其中123其实是不存在的一个文件夹,那么其实这句命令的意思就是对 linux.tar.gz这个文件进行重命名
总结一下:cp就是windows系统中的复制+粘贴,而mv就是剪切+粘贴。
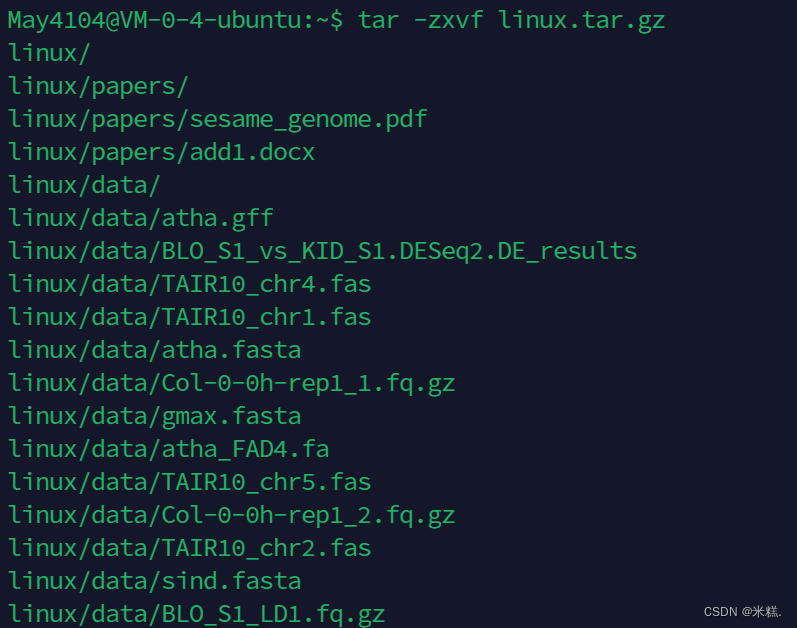
解压文件使用的命令是tar -zxvf 文件名,运行结果如图
查看某个文件夹中有哪些文件有两种方法,第一种是使用cd跳转到那个文件去,然后使用ls,ll等等命令来查看,第二种是使用ll 加文件夹名 直接查看,第一种方式就好像是我想要看看月球上有什么,我直接跑到月球上去看看,第二种方法是我在地球上拿一个望远镜来看看,是远程查看。
在Linux中查看文件内容仅限于文本文件,因为linux没有图形界面。什么是文本文件呢?是不是后缀是txt的?其实不一定,我们需要明确一点,Linux系统下的文件名中后缀不重要,不像windows系统中的文件名后缀那样重要。查看文件使用的命令是cat 加文件名,可以使用tab键来自动补齐。
cat 加文件名这种方法来查看文件仅限于查看小文件,如果一个文件的字节数很多,使用cat查看就会导致不停滚屏,不方便阅读,此时应该使用less命令来查看大文件,用法为less 加文件名,这时候会在一个新的窗口中打开。有时候会出现显示的"很乱"的情况,比如这样,这是我用less命令查看一个大文件的结果,这样露娜的原因是因为字体太大了,屏幕放不下,所以这一行的内容就跑到下一行去显示了,就显得很乱,要解决这个问题,只需要使用less -S即可
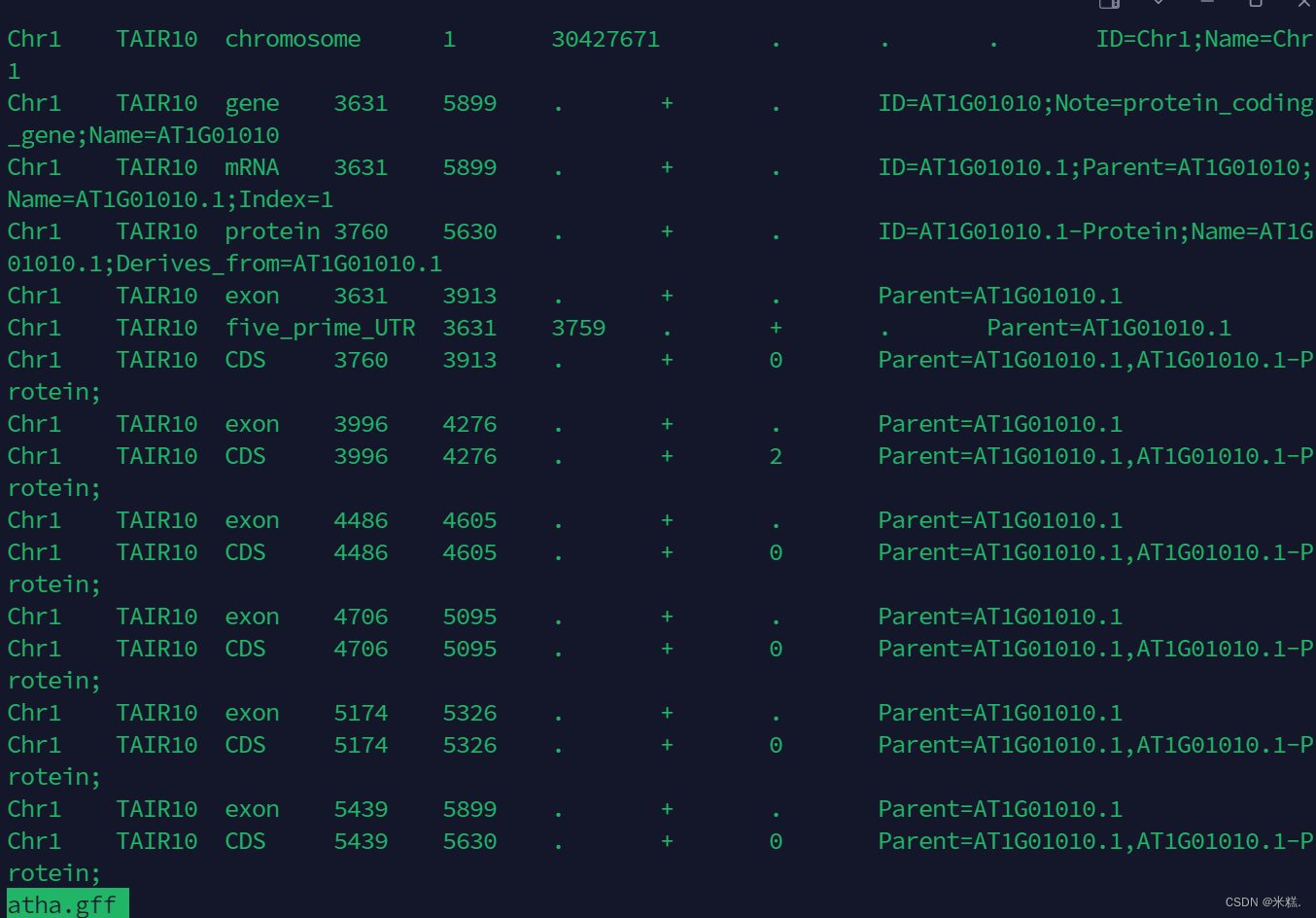
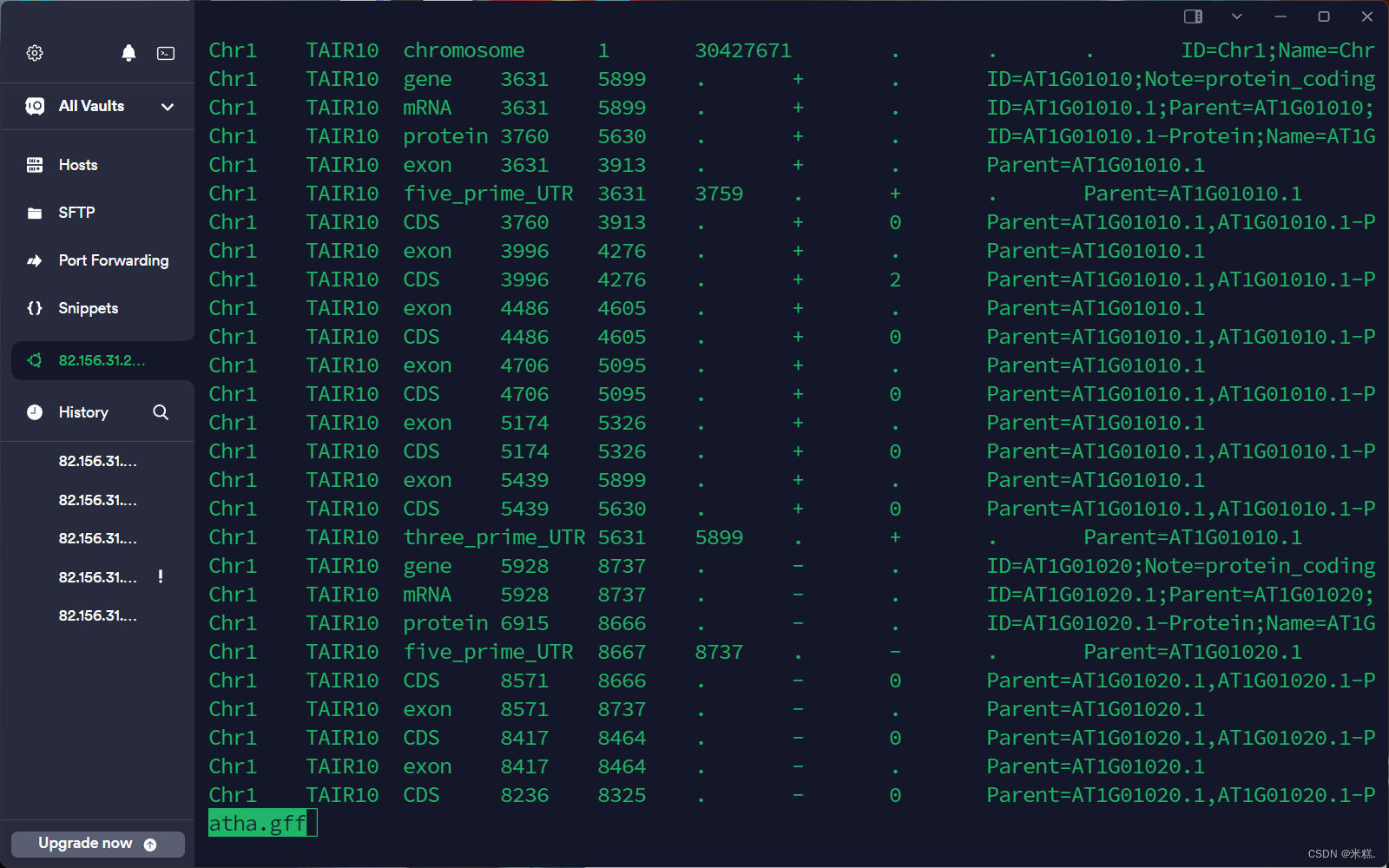
使用less -S atha.gff查看atha.gff这个文件的内容结果如图
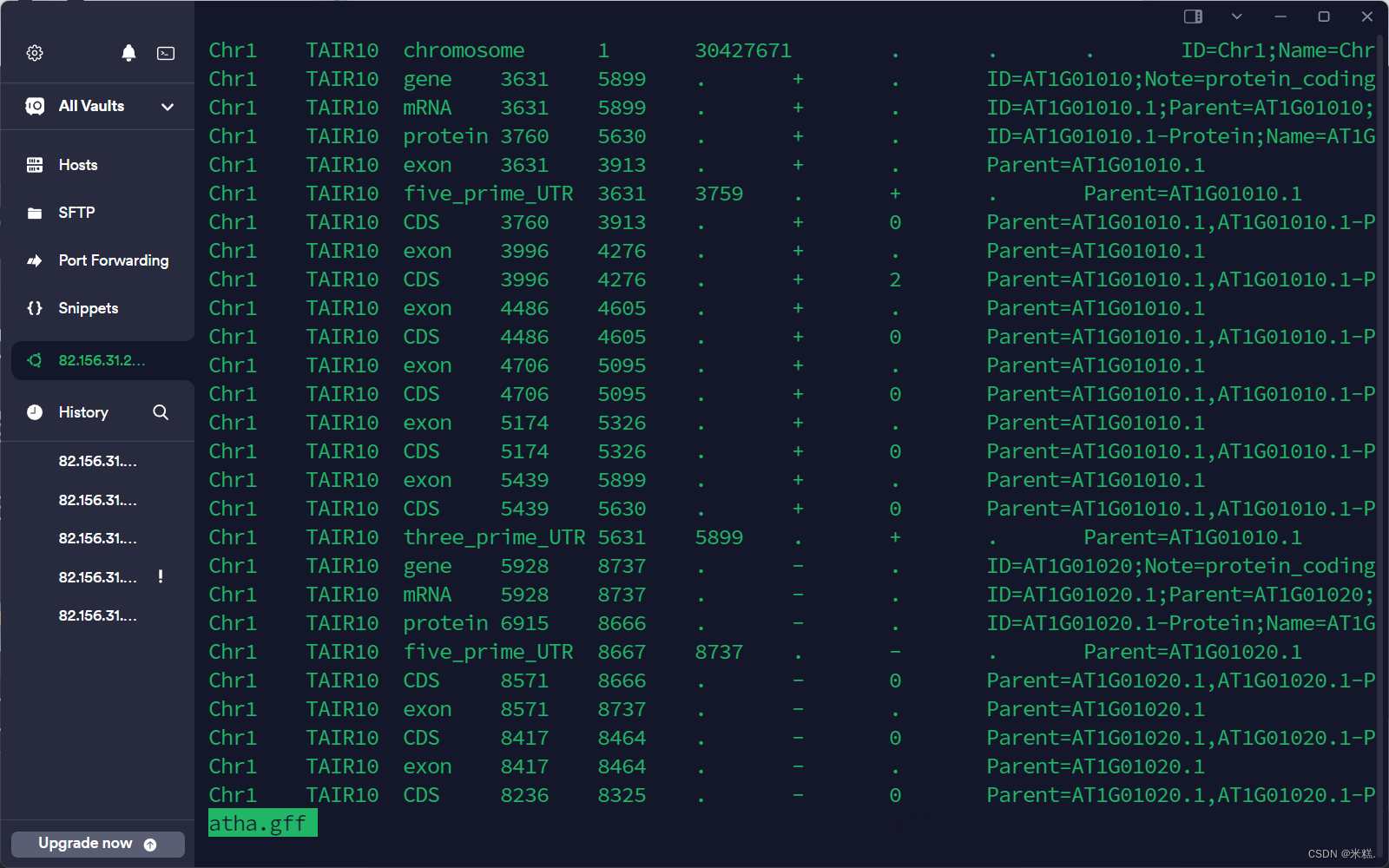
可以看到这个gff的文件里面有很多信息,比如第一列是位于哪个染色体上,第二列不重要,第三列是对应的基因,或者mRNA,蛋白质等等,第四列第五列是从哪个碱基开始,到哪个碱基结束。
如果我们想要查看这张表中某一个基因的信息,在打开这张表的基础上,按一下/,然后输入基因的名字(不一定是基因名,也可以是序列等别的信息)比如AT2G22890.1,结果就是这样
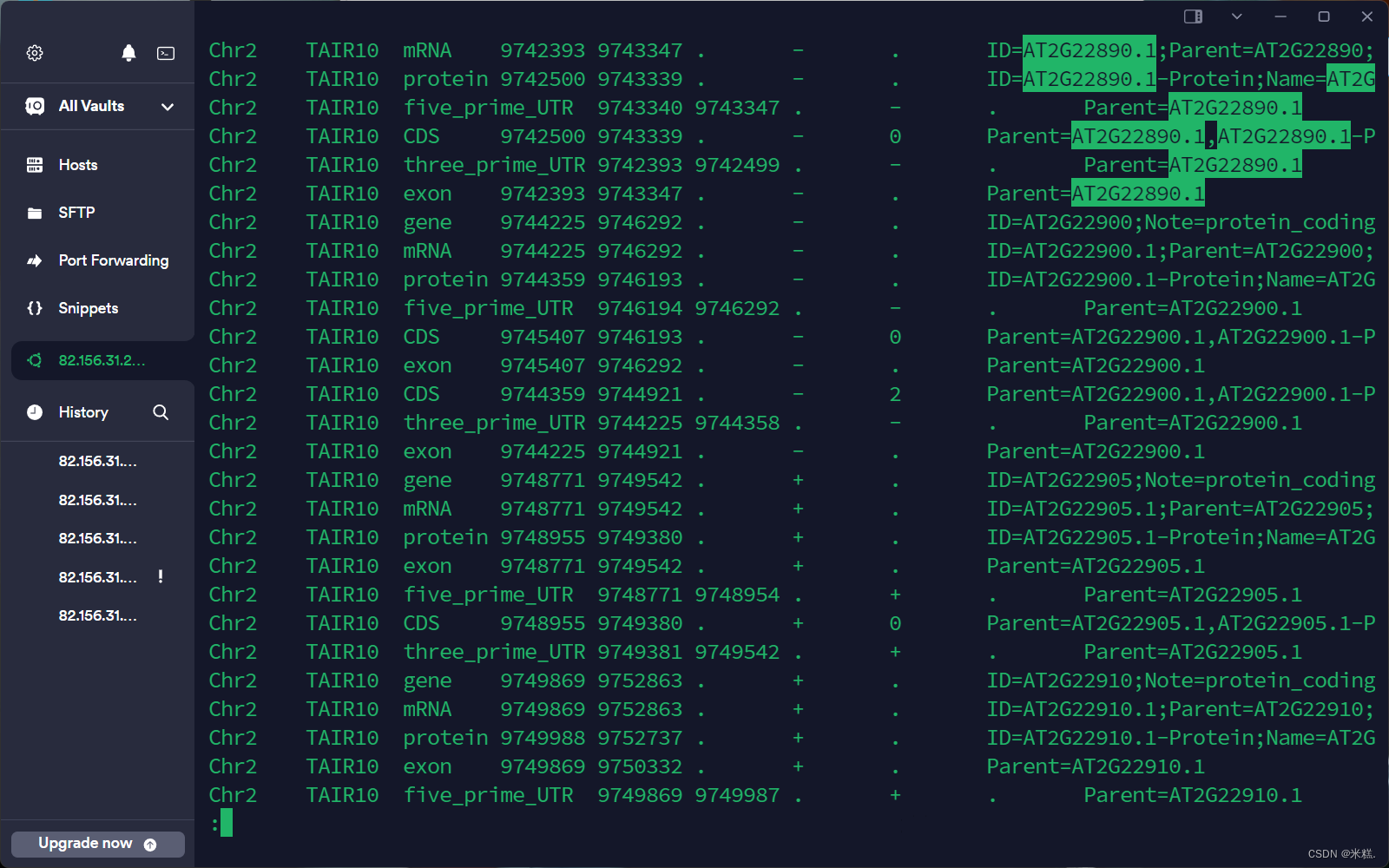
确实是只有AT2G22890.1这个基因的信息了
如果我们想要看到行号,可以用命令less -S -N,结果如图
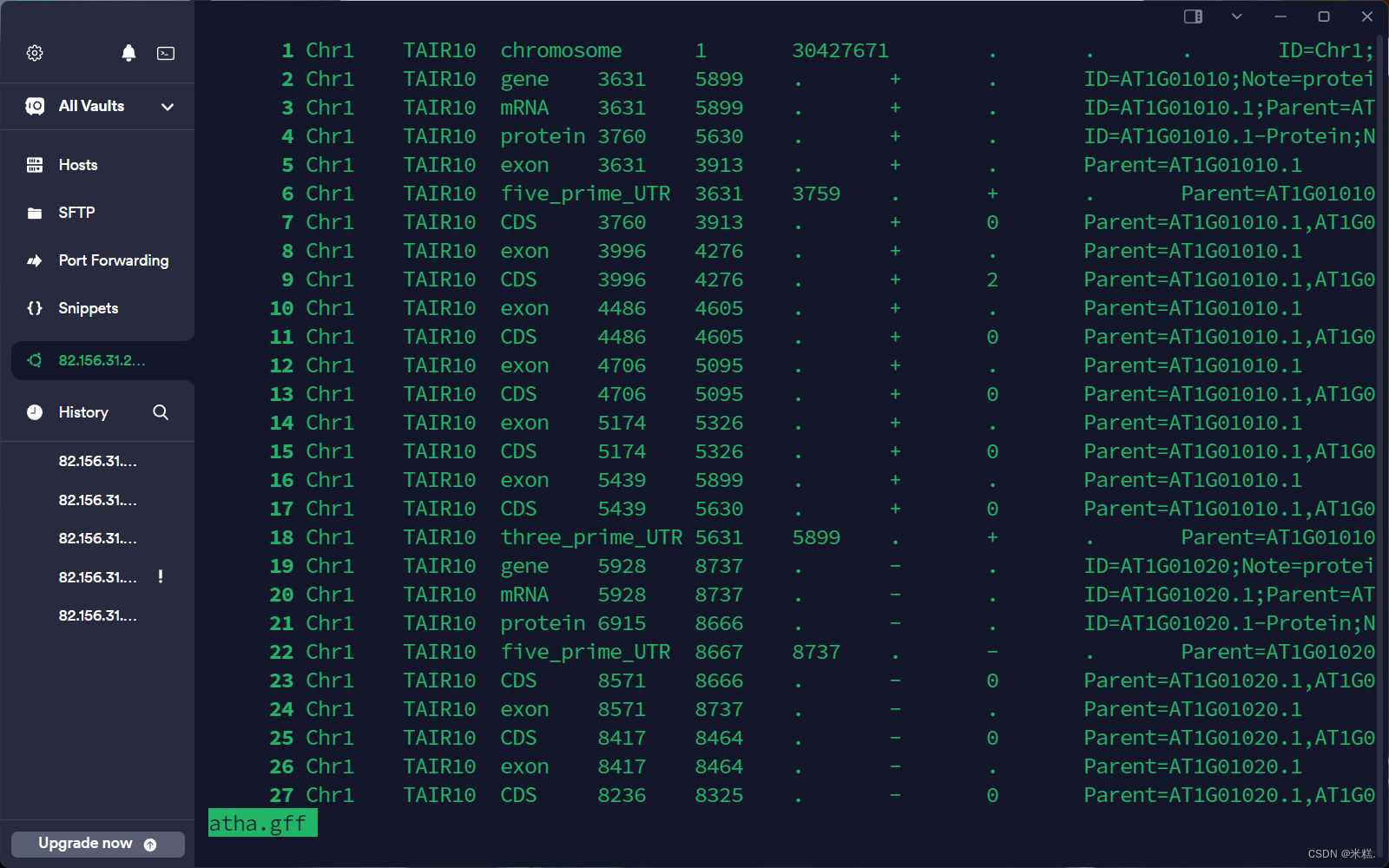
如果我们想看文件的前几行或者后几行,用到的命令分别是head和tail,默认是看前10行或者后10行,如果想要指定看多少行,可以写head -n 12 加文件名,表示要查看当前文件的前12行。
新建文件使用的命令是vi 加文件名,比如我输入vi lubenwei.txt就有了这样的一个界面
但是此时我们并不能对文件内容进行编辑,只是创建了一个文件,我们使用vi命令表示使用vim的编辑器来创建文件
当创建好了文件之后我们如果想要编辑文件内容应该先按一下i来进入编辑模式,可以发现此时的左下角已经变成了这样
我们就可以输入内容了。
要想退回到一般模式只需要按一下esc即可。一般模式的标志就是左下角什么也没有。
还可以输入相应的按键来进入命令行模式,比如先输入冒号,在输入wq就是保存并退出,可以发现此时已经有了我们刚才创建的文件,如果是不保存并退出就按了冒号之后用q!。有时候不加感叹号也表示不保存并退出,取决于服务器,所以我们使用vi命令创建文件之后如果想要退出,要么就按了冒号之后再按wq,要么就按了冒号之后再按q!
刚才我们已经创建了lubenwei.txt这个文件,那么此时离我们再次运行命令vi lubenwei.txt这个命令就会打开我们刚才创建并编辑过的文件。如图,这是我刚才输入的内容

一般模式下可以复制和粘贴,首先使用鼠标点击到某一行去,再输入yy就可以复制这一整行,然后再按i进入编辑模式,按p就可粘贴了。yy这个方式就是一次复制一行,但是如果我们想要一次复制好几行,比如三行,就按3y,如果要选择性的复制某些内容,就要按v先进入可视化模式,如图
然后按小键盘的上下键来选中要赋值的内容,之后按一个y就可以复制了,粘贴还是p
删除的命令逻辑和选中复制的命令类似,如果要删除光标所在的当前行,使用命令dd即可,如果要删除从光标开始的三行,就写3d,如果要选择性的删除某些内容,就先按v,然后小键盘上下左右键选中内容,然后按d即可。
搜索vi创建的某些内容先按/,然后输入要搜索的内容,如果要撤销上一步的操作,按u即可。
文本操作:
重定向命令是>,比如head -500 atha.gff > tmp表示把atha.gf这个文件的前500行拿出来放到临时文件tmp中去,atha.gf的内容长这样子,之后我们可以查看tmp的内容
运行less -S tmp结果如图
利用重定向">",我们可以完成下面的任务:
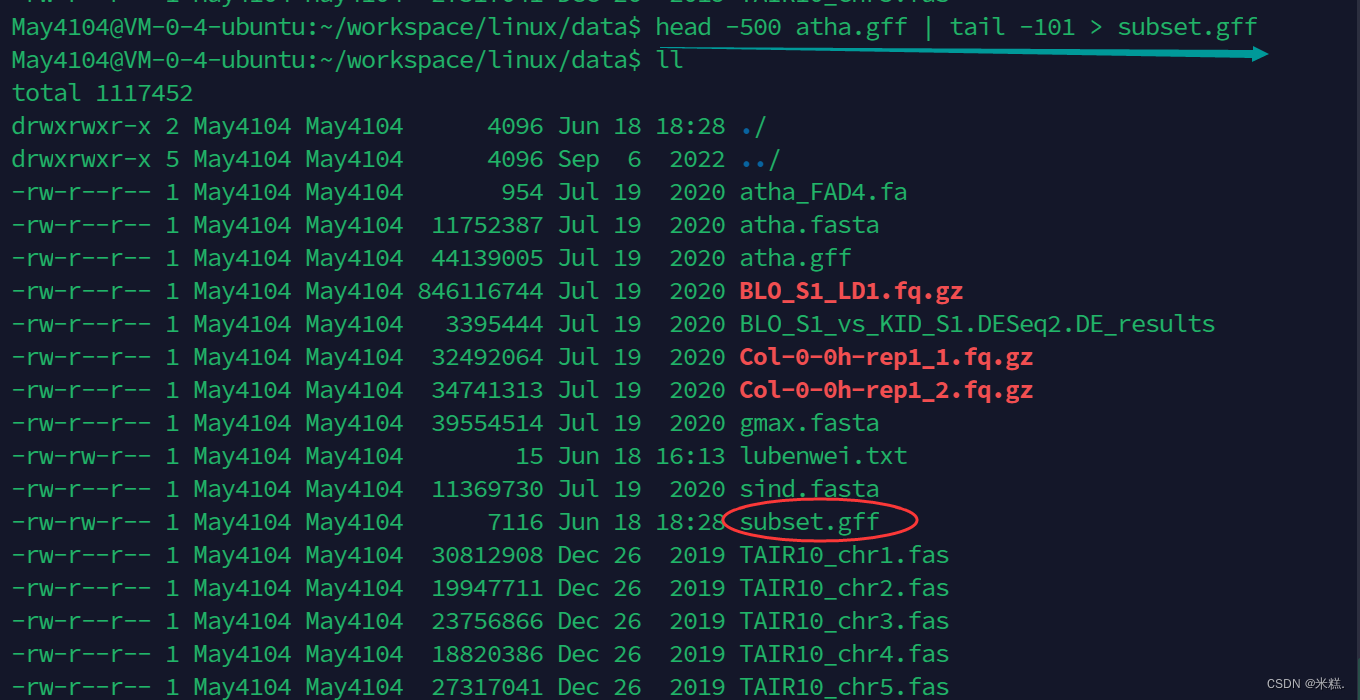
提取 atha.gff 的 400 到 500 ⾏,打印输出到 subset.gff
首先head -500 atha.gff > tmp提取出前500行放到tmp里面,然后再tail -100 tmp > subset.gff 取出tmp的后100行放到subset.gff 里面,看起来很合理,但是对吗?其实这样做就少了一行,因为我们这样操作最后subset.gff 里面的内容是第401行到500行的内容,但是题目要求是取出400到500行一共101行的内容,所以我们要写成tail -101 tmp > subset.gff,其实还有另一种更简洁的写法就是head -500 | tail -101 >subset.gff,其中用竖杠来代表临时前面的命令的结果(等价于上一种写法的tmp),后续的tail -101 tmp中的tmp也不用写了,会自动用|的内容也就是head -500 的结果来替换。结果如图,subset.gff也被成功创建了出来。
注:1.在linux中,|被称为管道。
2.重定向 > 的效果为覆盖,如果已经有名为tmp的文件,就会覆盖掉原本文件中的内容。如果要追加内容,请使用>>。
这种方法不仅简洁,而且不会生成中间文件tmp,缺点是可读性不好。
查看 atha.fasta ⽂件中有多少条序列
首先来看一下atha.fasta内容是什么。
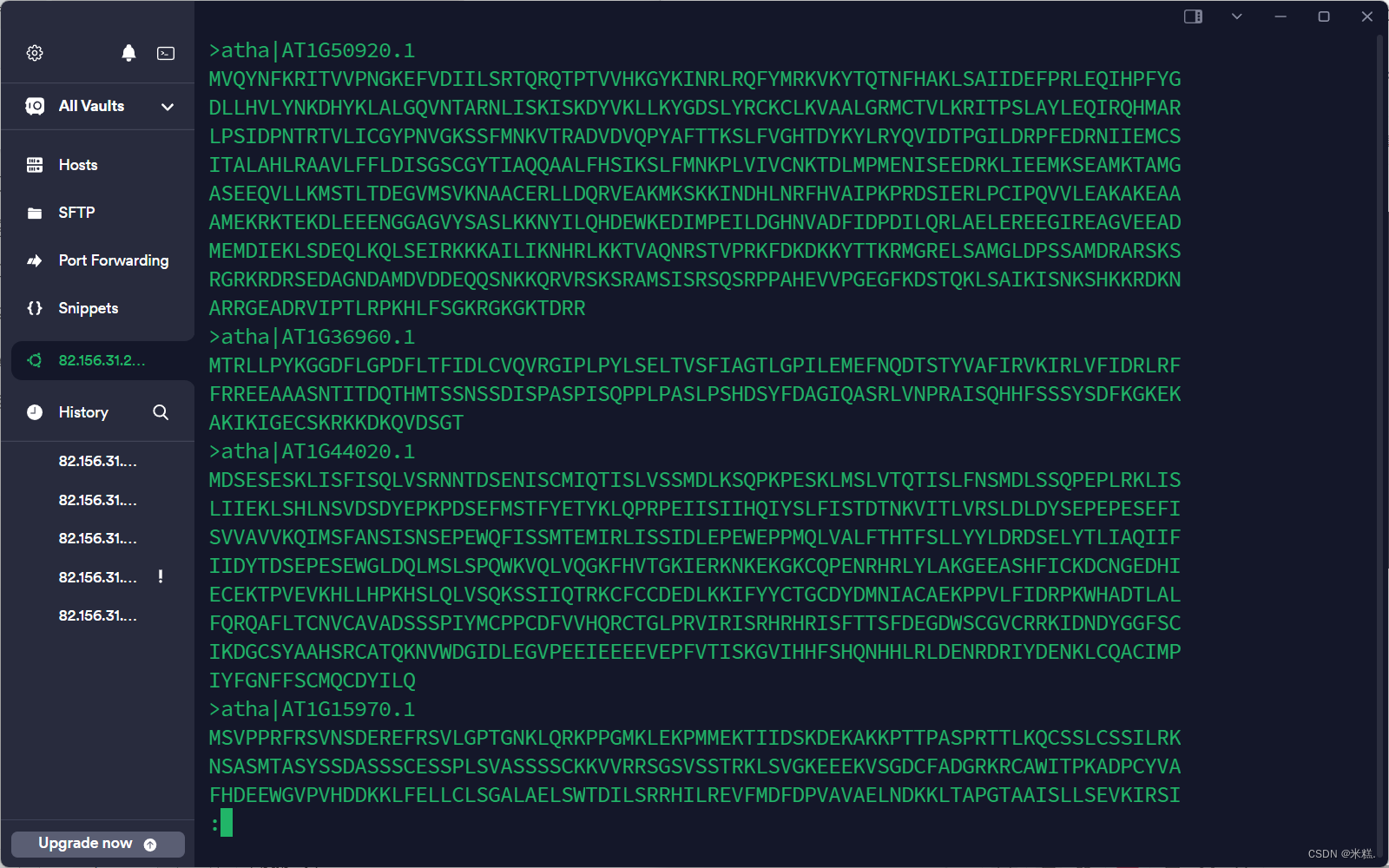
其中每一段是一个基因与他的序列,我们想要看看这个文件中有多少条序列其实只需要看看有多少个这样的"片段"就行了,每一个片段都有一个特征就是他们都由>开头,所以我们只需要统计有多少个>就行了。
首先使用命令grep ">" atha.fasta>tmp生成一个临时文件tmp,这个文件中包含所有含有">"的行,如图
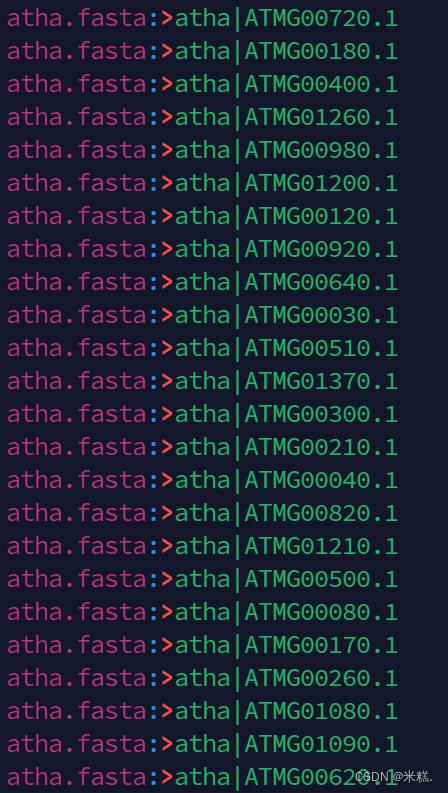
我们只需要统计这个文件有多少行就可以知道atha.fasta这个文件中有多少个基因了,统计某个文件的行数用的命令是wc,表示word count,输入wc tmp

结果显示有27416个基因。
练习:查看下表中有多少转录本的信息
其实就是把含有mRNA的行都提取出来放到一个文件里面然后统计行数就行了。用的命令就是grep "mRNA" atha.gff | wc。结果如图

再来看看有多少个基因:运行grep "gene" atha.gff | wc

发现基因的数量要比转录本的数量少。一个基因是可以转录出多个mRNA的,生物学上称为可变剪切。如果发现基因的数量和mRNA数量相等,那说明这个基因我们还没有研究明白,不知道他能转录多少种mRNA,所以我们称之为注释到了基因水平,如果像刚才那样我们发现基因的数量少于转录本的数量,说明我们已经研究明白了基因与mRNA的关系,称之为注释到了转录本水平。
实际上使用grep来跳出含有gene这个单词的行从而判断有多少个基因的方法不严谨,因为我们希望的只是第3列有gene这个单词,但是使用grep筛选的话即使别的列种有gene这个单词也会被算进去,正确的方法是使用命令awk来指定某一列筛选,比如我运行命令awk '3=="gene"' atha.gff \|less -S,语法为先写上awk,然后是单引号,单引号内是筛选的条件,其中3表示第三列,之后跟上要筛选的文件。结果如图
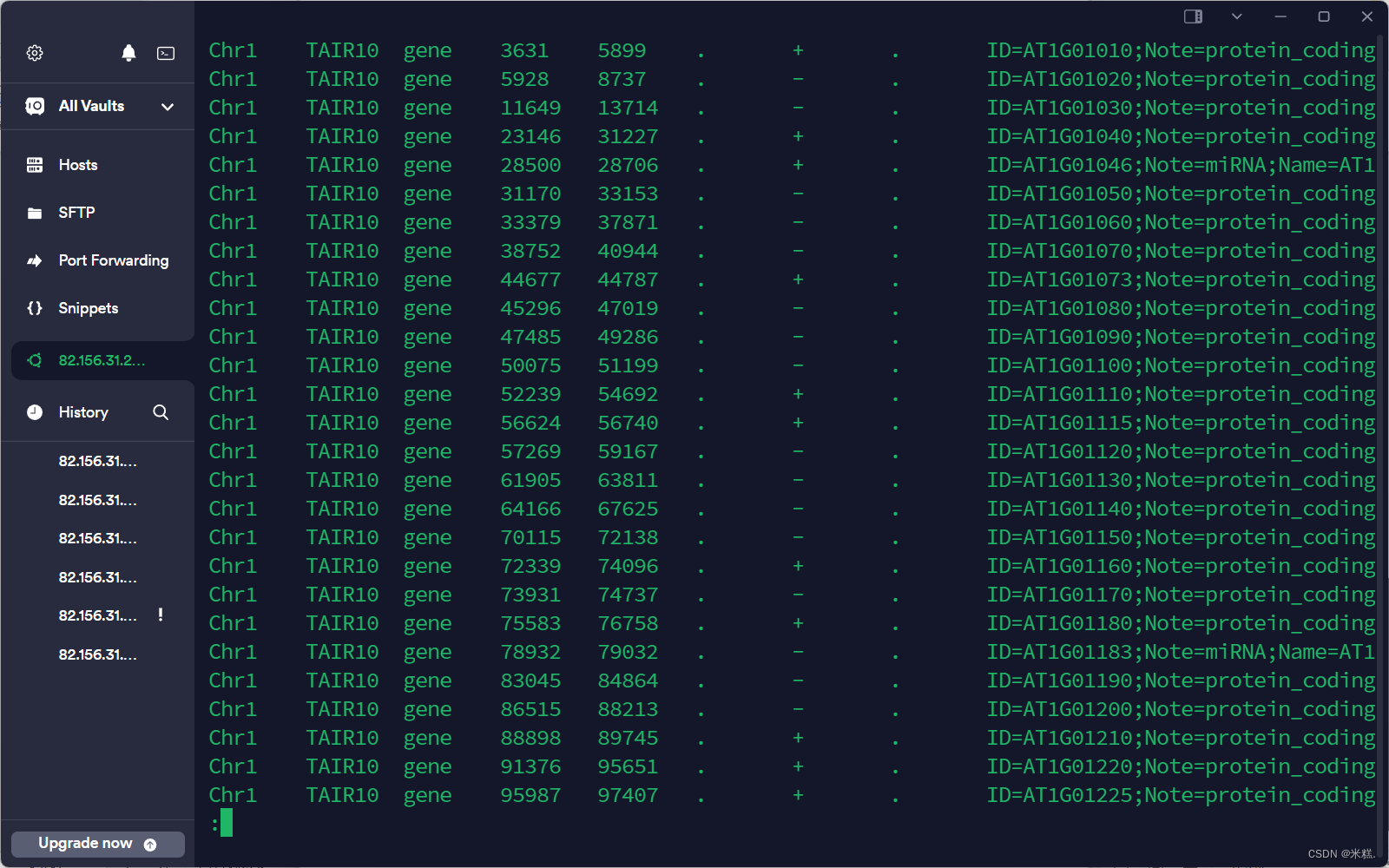
此时再用wc命令来统计awk '$3=="gene"' atha.gff |wc,结果如图

变成了两万多个基因。这种方法才是严谨的方法,说明刚才我们直接使用grep进行筛选的时候多算了很多行进去。
awk命令还可以把某一列内容打印出来,使用命令:
awk '{print$3}' 加文件名
接下来再看一个文件,名为BLO_S1_vs_KID_S1.DESeq2.DE_results
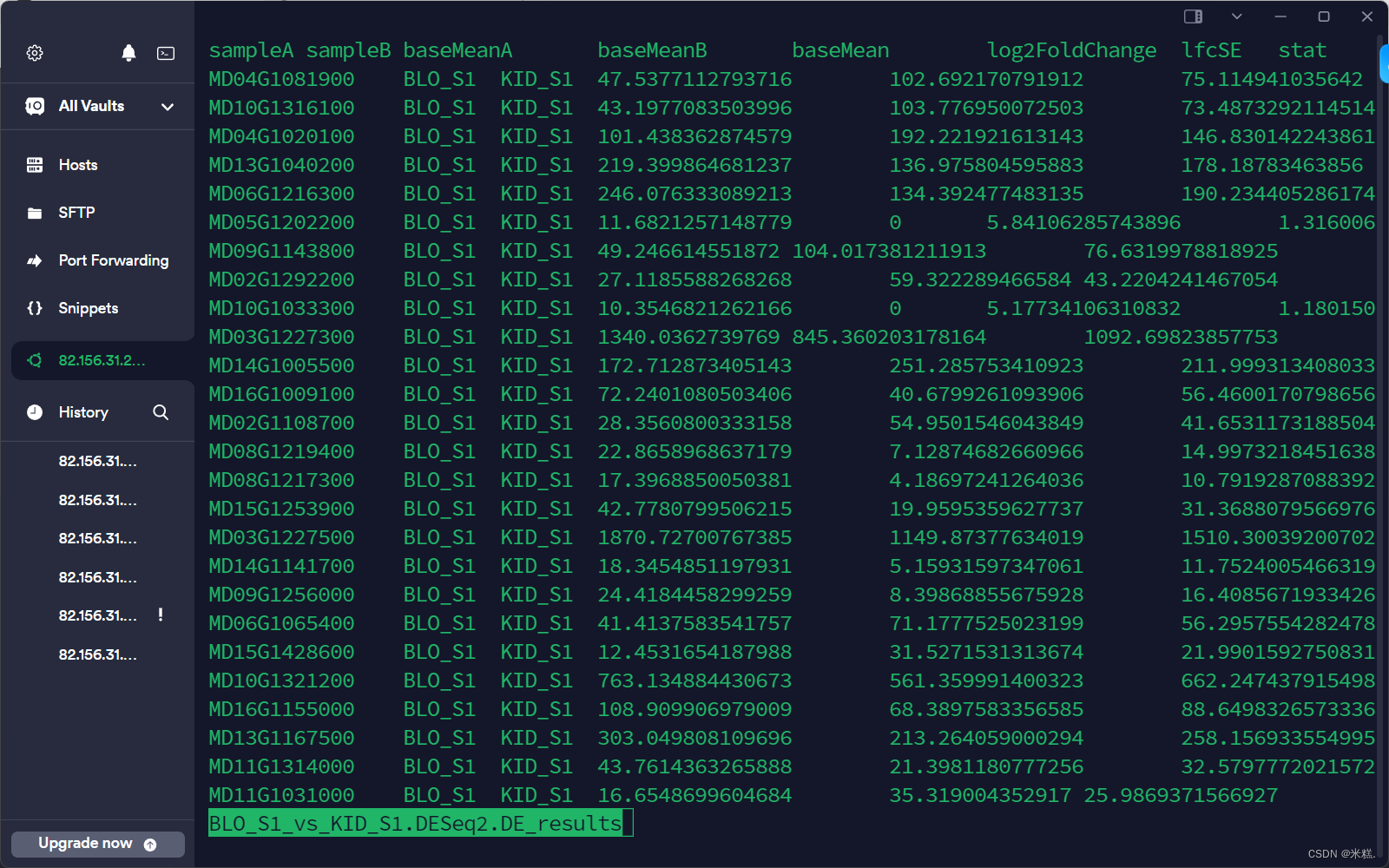
里面包含了差异分析的结果,比如logFC和P值等等,其中logF在第七列,P值在第11列,我想要挑选出同时满足logFC大于1和p值小于0.05的行,使用Linux中的awk命令也可以做到,写作:
wak '7\>1\&\&11
如果我想要删除BLO_S1_vs_KID_S1.DESeq2.DE_results的第一行,因为第一行是表头,不能参与运算,我可以直接vi BLO_S1_vs_KID_S1.DESeq2.DE_results打开这个文件,然后光标弄到第一行按dd就能删掉了。或者使用命令sed '1d' BLO_S1_vs_KID_S1.DESeq2.DE_results 也可以。两种方法等效。其中sed后面的单引号里面放1d表示删除第一行。
如果要筛选出绝对值大于1的行呢?在R语言中我们可以使用abs这个函数来求绝对值,但是在Linux中没有这个函数,但是却有sqrt表示开平方的操作,我们又知道x*x然后再开平方就是绝对值x,所以筛选出绝对值大于1的行就写作awk 'sqrt(7\*7)>1' 文件名
压缩和解压
压缩文件的常用命令为gzip,运行gzip 加文件名 这句命令之后会在当前工作目录下生成一个gz文件。
打包文件用的命令是tar
比如我想要把当前工作目录下圈起来的这几个文件打包成一个文件
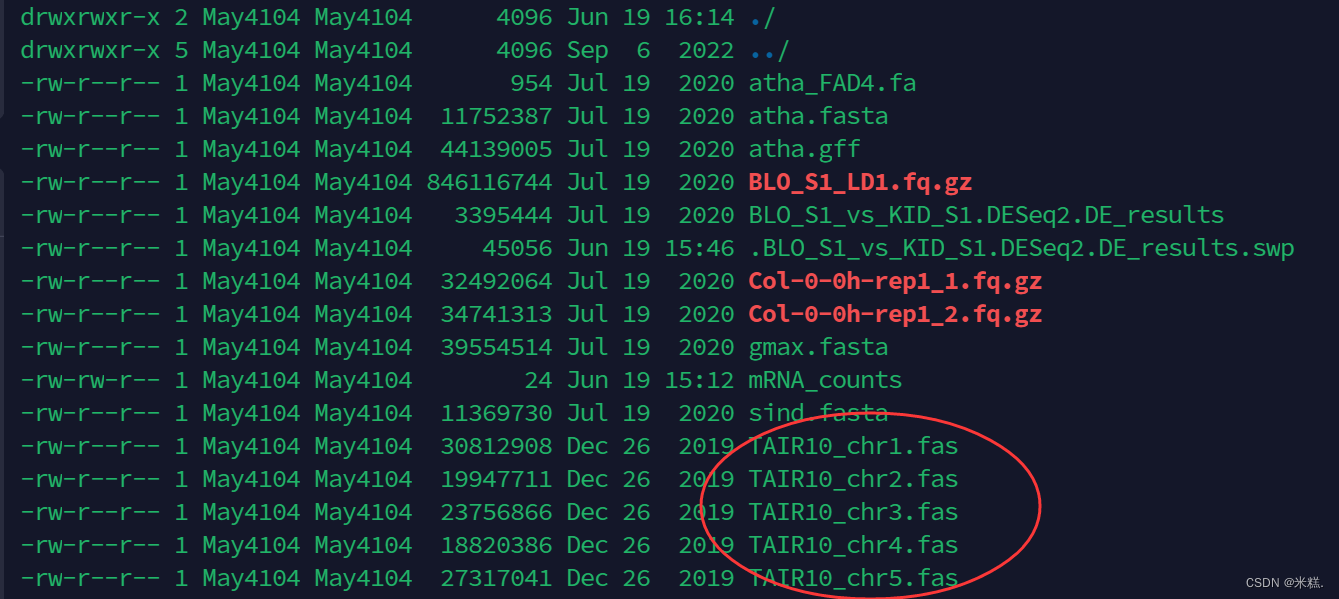
就运行这句命令:tar -c -f TAIR10.tar TAIR10_chr*.fas
其中tar是用于打包的命令,-c表示创建一个新文件,-f后面跟的是创建的文件的文件名,最后是要把哪些文件打包,当然可以把这几个文件名全复制下来写在这里,但是比较麻烦,我们发现这几个文件的文件名唯一不同的地方就是1 2 3 4这里的数字,我们使用*替代这个位置,*在这里是通配符的作用,表示只要含有TAIR10_chr什么什么.fas的文件名都要打包起来。运行之后再次查看当前工作目录结果如图
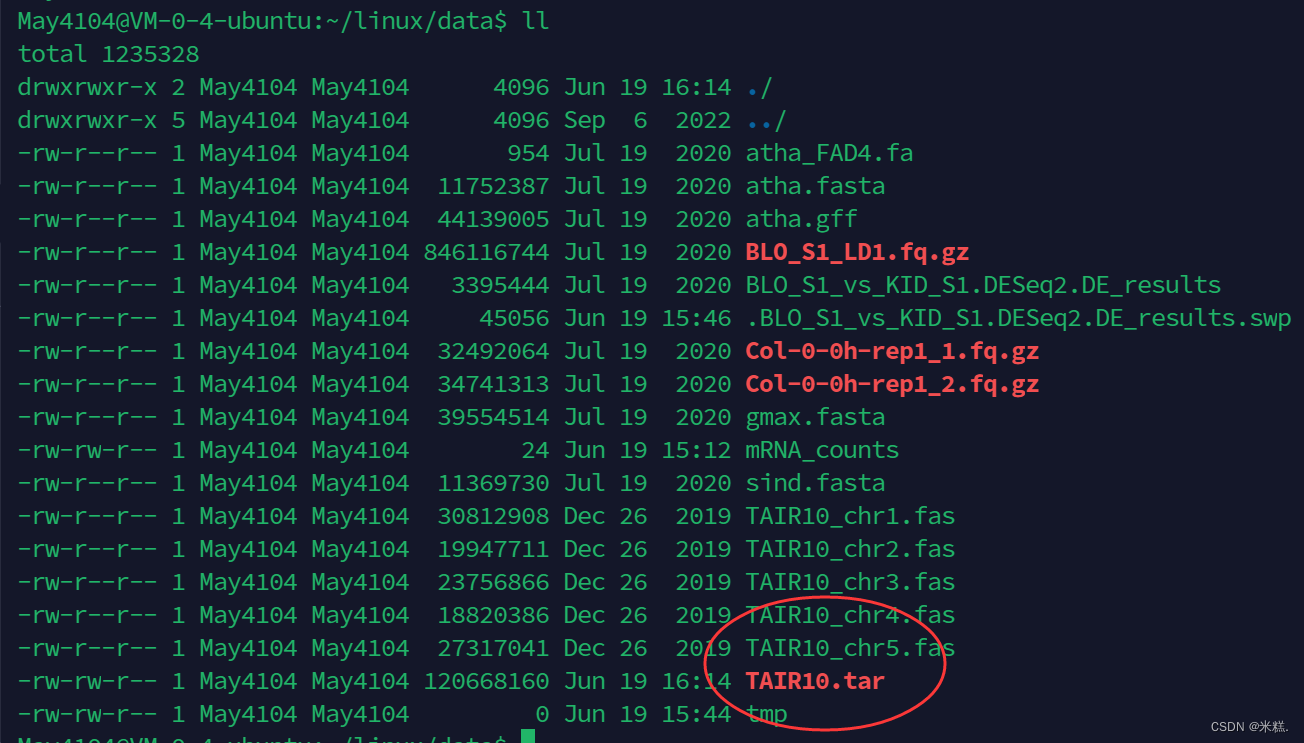
打包的文件已经被创建了出来。通常我们会把打包与压缩的操作同时进行,只要再tar命令中再加一个参数就-z就行了,压缩的过程会比较慢的,我们可以再加一个参数-v,这样每压缩完一个文件就会把这个文件显示在屏幕上,如图
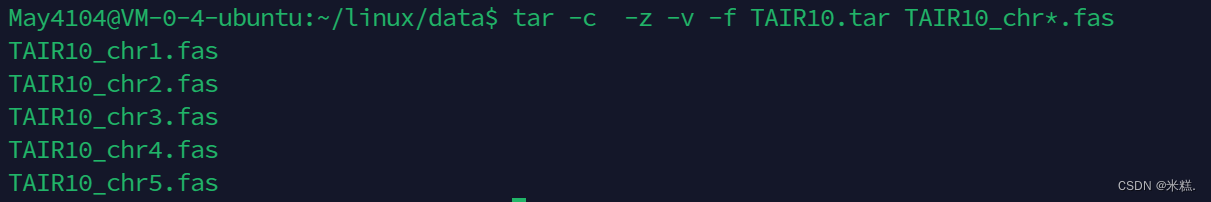
当然命令中的-可以省略,写成tar -czvf -文件名
解压缩还可以使用命令tar -cxcf -文件名
下载到服务器的软件要运行的话需要指定路径+软件名,比如我要运行fastp这个软件,就写成./fastp表示运行当前目录下的fastp软件,有时候会遇到这样的情况
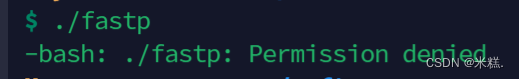
这是因为我们没有运行这个软件的权限,需要先使用命令chmod来设置权限,写a+x表示所有人都有这个权限,写u+x就是只有自己有权限,然后跟上软件的名字
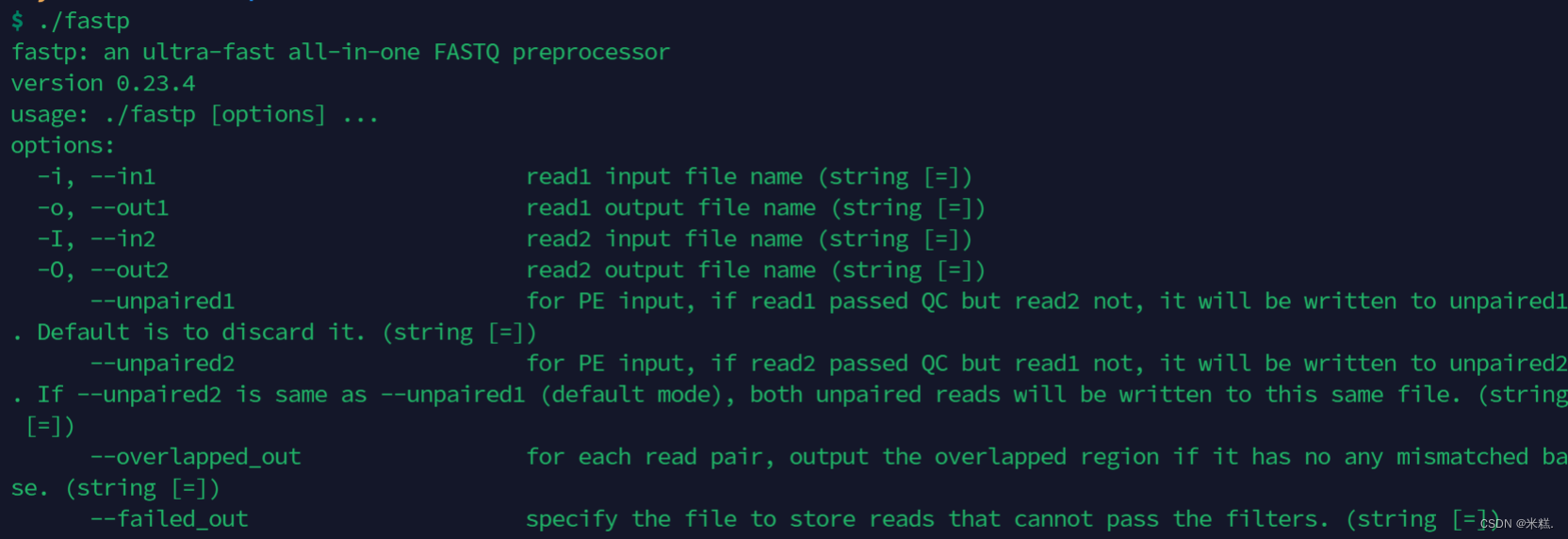
这样我们再次运行这个软件就可以了,出现了这个软件的帮助文档
添加环境变量
环境变量是Linux系统中自带的一个变量,名为$PATH,可以使用下面的命令来查看环境变量的内容

发现是好几个路径,注意这几个路径以冒号分割,分别是不同的路径,前面我们说了运行下载的软件需要指定路径,但是再运行一个名为htop的软件时,没有指定路径却能够正常运行,这是因为再运行指令'htop'的时候,系统自动把我们输入的htop这个命令替换成这样
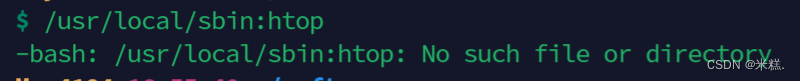
发现没有,接着又替换成$PATH的第二个路径

发现还是没有,再替换成第三个路径

一直遍历$PATH中的内容直到成功运行htop
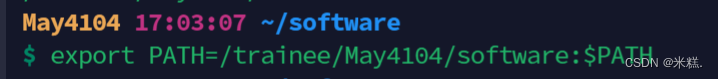
也就是说如果环境变量中有我们安装的软件所在目录的路径,我们就可以直接输入软件名来运行这个软件,在环境变量中添加内容用的命令是export PATH='路径':PATH ,注意等号的前后不要加空格,前面的PATH没有,后面的PATH有$。如图
这样环境变量就填加了这个路径,这个路径就是我们刚才安装fastp这个软件的路径,所以现在我们可以直接使用fastp这个命令来运行该软件。使用export命令在环境变量PATH中添加内容的时候将添加在最前面。千万不要忘记加后面的:PATH,否则意思就成了环境变量中只有本次添加的第一个路径了,这会导致很多命令比如ls都用不了了。
使用这种方式在PATH变量中添加内容只对当前会话生效,如果我们退出之后再次登陆就会发现PATH中还是那些内容,想要让我们添加的内容永久生效,需要把这个命令添加到.bashrc这个文件中,这个文件在家目录下,找到之后运行命令vi .bashrc,在文件最下面输入
export PATH=/trainee/May4104/software:PATH之后按冒号再按wq保存并退出,这样即使结束本次会话,下次登录该服务器也能直接使用软件名(比如我们刚才的fastp)来运行软件了。如果用这种方式来修改环境变量的时候忘记了后面的:PATH,后果就比较严重了,我们可能会想,只要再次vi .bashrc把刚才那句命令删掉不就行了?但是实际操作的时候我们发现vi这个命令不能用了,此时我们只能记住vi这个命令的绝对路径,从而使用绝对路径来使用vi这个命令,vim的绝对路径是user/bin,使用user/bin vim就能运行vim了。
使用which '软件名' 将返回软件装在了哪里,这可以辅助我们添加环境变量的内容。
.bashrc这个文件是一个系统的配置文件,启动系统的时候自动加载,前面我们提到过一个起别名的命令alias,比如我写
alias le=less- a,那么le这个命令就相当于less-a了,但是这样改也是只在本次会话中生效,当我们退出之后再次连接服务器就没用了,如果想要让别名永久生效,只需要像刚才修改环境变量那样把起别名的命令行写到.bashrc这个文件中保存退出即可。