细胞通讯分析是一种研究不同细胞类型之间如何通过信号分子(如配体和受体)进行相互交流和调控的分析方法。它在揭示细胞间相互作用的机制,理解组织和器官如何协调运作方面具有重要意义。
细胞通讯分析的主要内容如下:
-
配体-受体相互作用: 每个细胞类型都会表达特定的配体和受体。细胞通讯分析通过研究这些配体和受体之间的相互作用,来了解细胞是如何"交流"的。例如,一个细胞释放的配体可以与另一种细胞的受体结合,从而触发一系列细胞内的信号传导事件。
-
信号通路活性: 通过分析哪些信号通路在不同细胞类型之间被激活,研究者可以了解哪些信号通路在特定生物学过程中起作用,如细胞分化、免疫反应或疾病状态等。
-
细胞间网络构建: 细胞通讯分析通常会构建一个细胞间的网络图,显示不同细胞类型之间的相互作用方式。这个网络可以帮助识别主要的信号发送者和接收者,以及哪些细胞在组织中起主导作用。
-
定量分析: 通过计算细胞通讯的强度(如配体-受体结合的概率),可以定量分析不同细胞类型之间的通讯强度。这可以揭示在特定条件下,哪种细胞类型在通讯中占主导地位。
-
功能富集分析: 细胞通讯分析还可以与功能富集分析结合使用,探讨哪些生物学功能或通路受到特定细胞通讯的调控。
常用工具:
1、CellChat:这是一个常用的 R 包,可以识别和量化不同细胞类型之间的通讯,并提供丰富的可视化工具。
2、CellPhoneDB:另一个广泛使用的工具,专注于基于配体-受体相互作用的细胞通讯分析。
本次主要来学习一下cellchat这个工具。
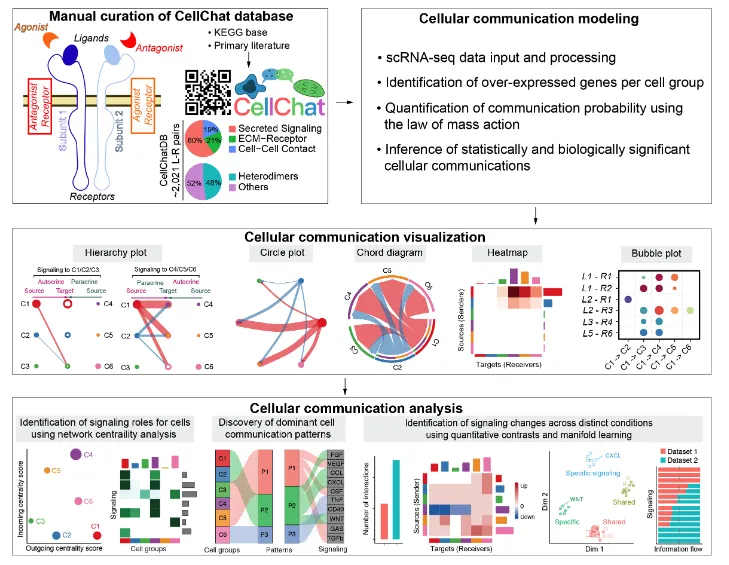

CellChatDB v2数据库中包含了丰富的先验信息:
1、数据库内容来源于已知文献的人和小鼠的配-受体相互作用信息。
2、这些信息包含了大约3300个经过验证的分子互作信息(~40%是自分泌/旁分泌,~17%是细胞外基质-受体,~13%是细胞-细胞间接触,~30%是非蛋白质信号)
3、相比于v1版本, CellChatDB v2增加了1000多种的蛋白质/非蛋白质的互作信息,比如代谢和突触信号传导。
4、对于数据没有直接作用的关键分子,该工具还能通过分析这些分子的关键介质/酶的情况来预测配-受体表达情况进而预测潜在通讯(这个是对~30%的非蛋白信号互作信息来源的解释)。
5、这个版本还增加了更多的附加注释信息:比如UniProtKB关键字(包括生物过程、分子功能、功能类别、疾病等)、亚细胞位置以及与神经递质的相关性。
6、人和小鼠的配受体库是不一样的,需要自行设定:CellChatDB.human/CellChatDB.mouse。

关于数据输入和准备的流程里开发者提供了不同数据的处理方式,包括conut matrix,seruat对象,SingleCellExperiment对象,Anndata对象等。
详细准备流程可以查看github官网,链接附在参考资料中,笔者这里使用的是Seurat对象。
步骤流程
1.导入
rm(list = ls())
library(CellChat)
library(Seurat)
library(qs)
library(patchwork)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
load('scRNA.Rdata')
# check一下
DimPlot(scRNA,pt.size = 0.8,group.by = "celltype",label = T)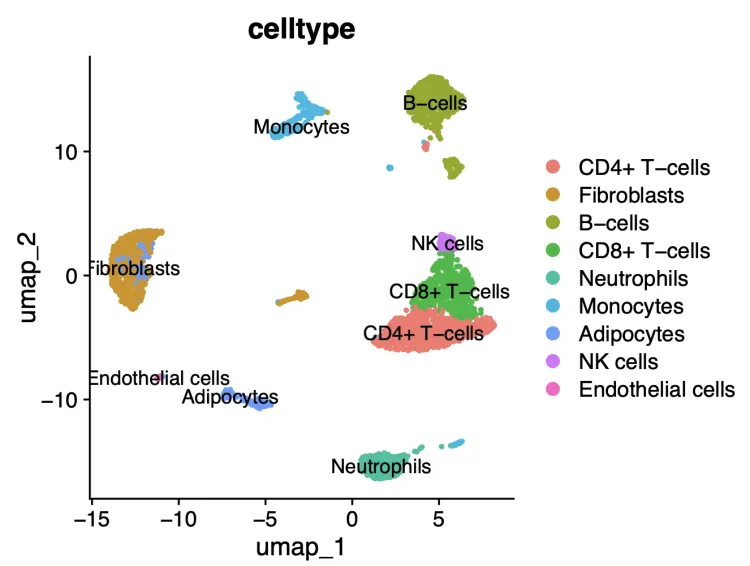
2.数据预处理
CellChat需要两个用户输入:一个是细胞的基因表达数据,另一个是用户分配的细胞标签。
对于基因表达数据矩阵,行为基因,列为细胞。
CellChat需要数据归一化数据。如果是count数据,里面内置了功能可以进行处理转换(但一般不用count,使用归一化数据)。
建议使用前先捋顺自己的数据。
data.input <- GetAssayData(scRNA, layer = 'data')
meta <- scRNA@meta.data[,c("orig.ident","celltype")]
colnames(meta) <- c("group","celltypes")
table(meta$celltypes)
meta$celltypes <- gsub(" cells|-cells", "", meta$celltypes)
table(meta$celltypes)
# Adipocytes B CD4+ T CD8+ T Endothelial Fibroblasts
# 244 667 1007 547 28 762
# Monocytes Neutrophils NK
# 311 378 87
# 建议提前对celltype进行排序~
identical(rownames(meta),colnames(data.input))
celltype_order <- c("CD4+ T","CD8+ T","B","Adipocytes","Endothelial",
"Fibroblasts","Monocytes","Neutrophils","NK")
meta$celltypes <- factor(meta$celltypes ,levels = celltype_order)
# 根据 meta$celltypes 的顺序进行排序
ordered_indices <- order(meta$celltypes)
# 对 meta 和 data.input 进行排序
meta <- meta[ordered_indices, ]
data.input <- data.input[, ordered_indices]
identical(rownames(meta),colnames(data.input))3.创建Cellchat对象
# 构建cellchat
cellchat <- createCellChat(object = data.input,
meta = meta,
group.by = "celltypes")
levels(cellchat@idents)
# [1] "CD4+ T" "CD8+ T" "B" "Adipocytes" "Endothelial" "Fibroblasts"
# [7] "Monocytes" "Neutrophils" "NK" 4.设置配体-受体相互作用数据库
CellChatDB <- CellChatDB.human # use CellChatDB.mouse if running on mouse data
showDatabaseCategory(CellChatDB)
dplyr::glimpse(CellChatDB$interaction)
# 使用CellChatDB的中特定的数据库进行细胞-细胞通信分析
# 示例中使用了Secreted Signaling
# CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling", key = "annotation")
# Only uses the Secreted Signaling from CellChatDB v1
# CellChatDB.use <- subsetDB(CellChatDB, search = list(c("Secreted Signaling"), c("CellChatDB v1")), key = c("annotation", "version"))
# 使用所有CellChatDB数据进行细胞-细胞通信分析。
CellChatDB.use <- CellChatDB
# 在构建的cellchat中设定需要使用的数据库
cellchat@DB <- CellChatDB.use5.预处理细胞-细胞通讯分析的表达数据
cellchat <- subsetData(cellchat)
# future::plan("multisession", workers = 1) # do parallel
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
# 默认情况下,cellchat使用object@data.signaling进行网络推断
# 此外提供了projectData函数,将基因投射到PPI,开发者说PPI并不会或导致很少的伪通讯
cellchat <- projectData(cellchat, PPI.human)
如何推断细胞间的通讯信息:
该工具可识别纳入分析数据的细胞群中存在哪些过表达的配体或者受体 ,然后去识别这些过表达配体或者受体与他们各自互作分子的相互作用信息。可以将基因表达数据映射到蛋白-蛋白互作网络上。
如何预测细胞间通讯情况:
1、Cellchat给每个交互作用情况一个概率并进行排列检验(permutation test)来推断生物学意义上的细胞通讯。
2、CellChat通过使用质量定律将基因表达与信号转导配体、受体及其辅因子之间相互作用的先验已知知识相结合,对细胞间通讯的概率进行建模。其中质量定律(law of mass action)的含义是:在细胞-细胞通信的背景下,质量作用定律被用来描述配体(ligand)与受体(receptor)之间相互作用的概率。具体来说,当一个配体与一个或多个受体结合时,信号的传递效率可以被认为是与配体和受体浓度的乘积成正比。这意味着,配体和受体的表达水平越高,细胞间信号传递的可能性越大。
3、配体-受体对的数量是基于每个细胞组平均基因的表达情况 。默认采用的计算方式是"trimean",这是一种严格的方法。如果想要宽松一些就可以在computeCommunProb中设定type = "truncatedMean" 和 trim = 0.1,比如computeAveExpr(cellchat, features = c("CXCL12","CXCR4"), type = "truncatedMean", trim = 0.1)。
4、假设分析未对细胞进行细分的单细胞转录组,在假设丰富的细胞群比稀少的细胞群更倾向于集体发送更强的信号的情况下,CellChat还可以在概率分析中考虑每个细胞群中细胞比例的影响(这个可能是指没有进行明确定义细胞类型的数据可以这么分析)
6.预测细胞-细胞通信网络
# 该分析的关键参数是类型,即计算每个细胞组的平均基因表达的方法。
# 默认情况下,type = "triMean",产生较少但更强的交互。
# 当设置 type = "truncatedMean" 时,应对trim设置一个值,进而产生更多交互。
cellchat <- computeCommunProb(cellchat, type = "triMean",raw.use = TRUE)
# 如下
# computeAveExpr(cellchat, features = c("CXCL12","CXCR4"),
# type = "truncatedMean",trim = 0.1)
# min.cells是设置阈值,最小是需要10个细胞参与通讯推断(可以自定义)
cellchat <- filterCommunication(cellchat, min.cells = 10)7.在信号通路水平上推断细胞间通讯
# CellChat通过汇总与每个信号通路相关的所有配体-受体相互作用的通信概率来计算信号通路水平上的通信概率。
# NB:推断的每个配体-受体对的细胞间通信网络和每个信号通路分别存储在槽'net'和'netP'中。
cellchat <- computeCommunProbPathway(cellchat)
# 计算聚合细胞-细胞通信网络
# 互作网络整合,可以设置soure和target,不设置就是默认全部
cellchat <- aggregateNet(cellchat)
#数据提取,subsetCommunication函数
df.net <- subsetCommunication(cellchat)
qsave(cellchat,"cellchat.qs")
save(df.net,file = "df.net.Rdata")
head(df.net)df.net文件中提取了不同细胞类型所检测到的互作信息。
8.可视化细胞-细胞间通信网络。
circle图展示互作数目
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = groupSize,
weight.scale = T, label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize,
weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")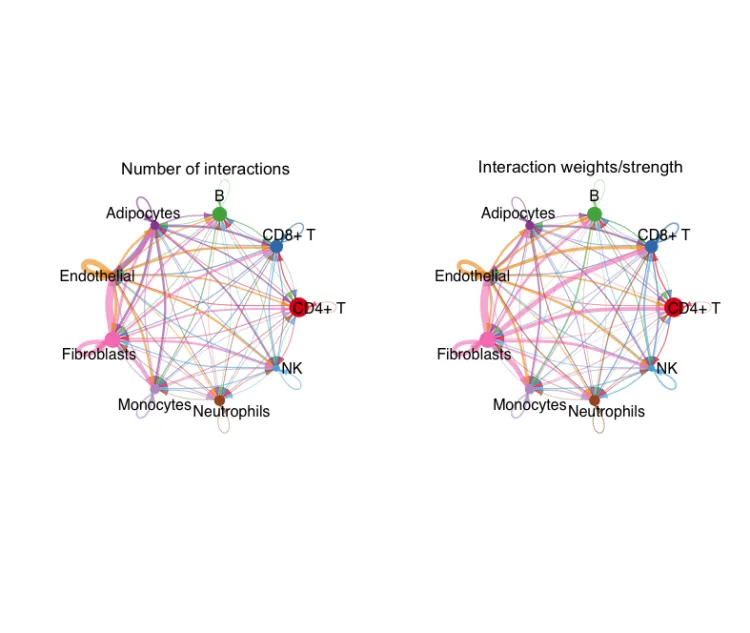
这两张图分别展示了互作的数量和互作的权重。其中每个颜色代表了不同的细胞,箭头代表了顺序,线的粗细代表了数量/权重。
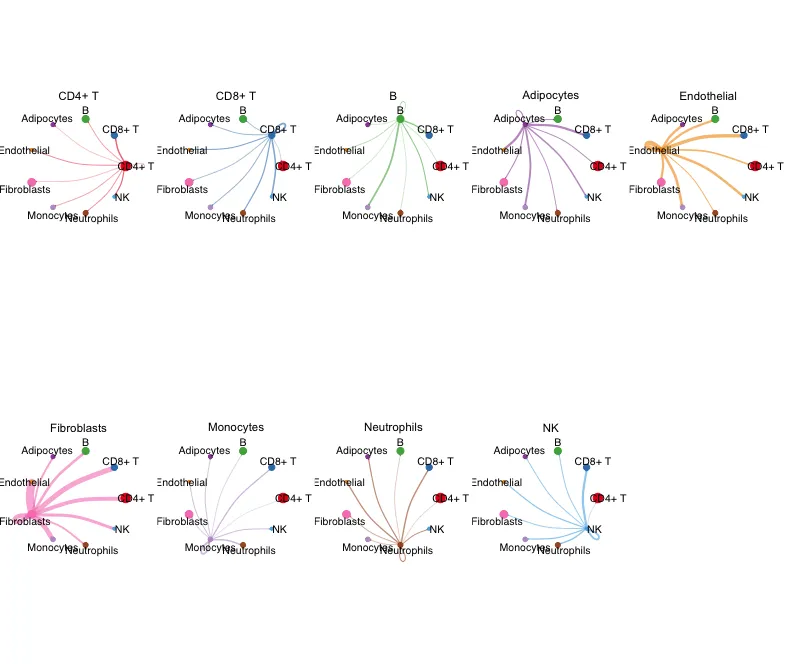
9.细分亚组的circle图
展示每一个celltype作为source与其他celltype的权重互作情况
mat <- cellchat@net$weight
par(mfrow = c(2,5), xpd=TRUE,mar = c(1, 1, 1, 1))
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(mat2, vertex.weight = groupSize,
weight.scale = T, arrow.size=0.05,
arrow.width=1, edge.weight.max = max(mat),
title.name = rownames(mat)[i]
)
}

简单一句话:接下来可以各种可视化。

层次结构图(Hierarchy plot) :该图有两个部分组成,左边部分显示对研究者感兴趣的细胞组(vertex.receiver)的自分泌/旁分泌信号传导,右边则显示数据集中其他次要感兴趣细胞的自分泌和旁分泌信号传导。
和弦图(Chord diagram) :提供了netVisual_chord_cell 和 netVisual_chord_gene 两个功能。netVisual_chord_cell 用于可视化不同细胞组之间的细胞间通讯;etVisual_chord_gene 用于可视化由多个配体受体或信号通路介导的细胞间通讯。
对边的颜色/权重、节点颜色/大小/形状的解释:边的颜色与信号发送源的颜色一致,边的权重与交互强度成正比。
10.层次结构图/circle图/弦图/热图
cellchat@netP$pathways
levels(cellchat@idents)
vertex.receiver = seq(1:3) # a numeric vector
pathways.show <- "COLLAGEN"
# Hierarchy plot
netVisual_aggregate(cellchat, signaling = pathways.show,
vertex.receiver = vertex.receiver,layout= "hierarchy")
# vertex.size = groupSize)
# circle plot
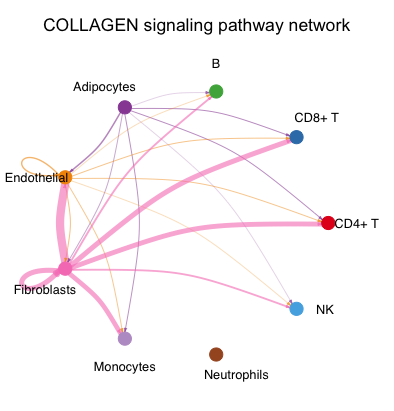
netVisual_aggregate(cellchat, signaling = pathways.show,layout = "circle")
# Chord diagram
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord")
# 分组弦图
levels(cellchat@idents)
group.cellType <- c(rep("T/B", 3),
"Adipocytes","Endothelial","Fibroblasts",
"Monocytes","Neutrophils", "Neutrophils")
names(group.cellType) <- levels(cellchat@idents)
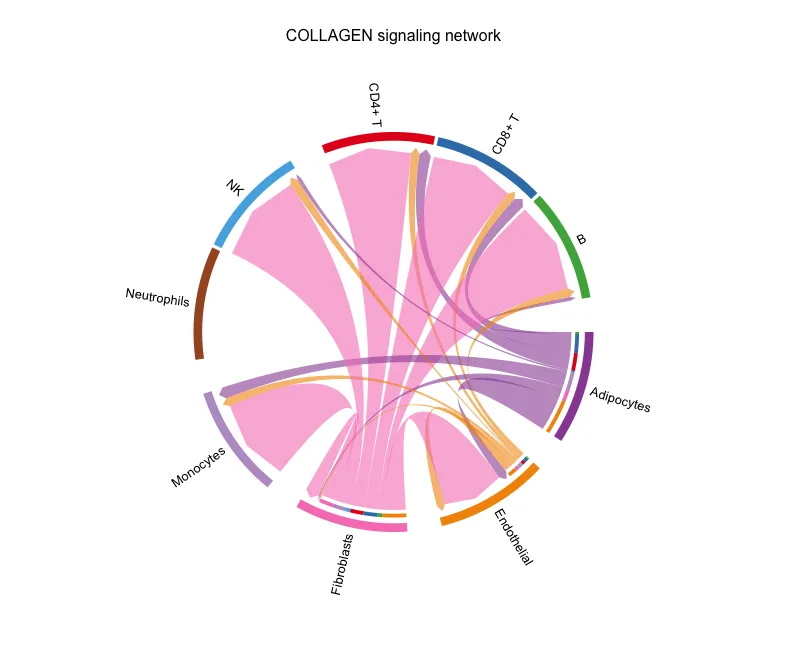
netVisual_chord_cell(cellchat, signaling = pathways.show,
group = group.cellType,
title.name = paste0(pathways.show, " signaling network"))
# heatmap
par(mfrow=c(1,1))
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")层次结构图
vertex.receiver设定了source的细胞类型(实心圆圈),空心圆圈代表target。左半边图片先把代表vertex.receiver的圆圈放在了中间,显示了不同细胞类型对这CD4+T,CD8+T和B细胞的作用情况,右半边图片把代表一起细胞的源泉放在了中间,显示了不同细胞类型对其余细胞的作用情况,线的粗细代表互作的强度。
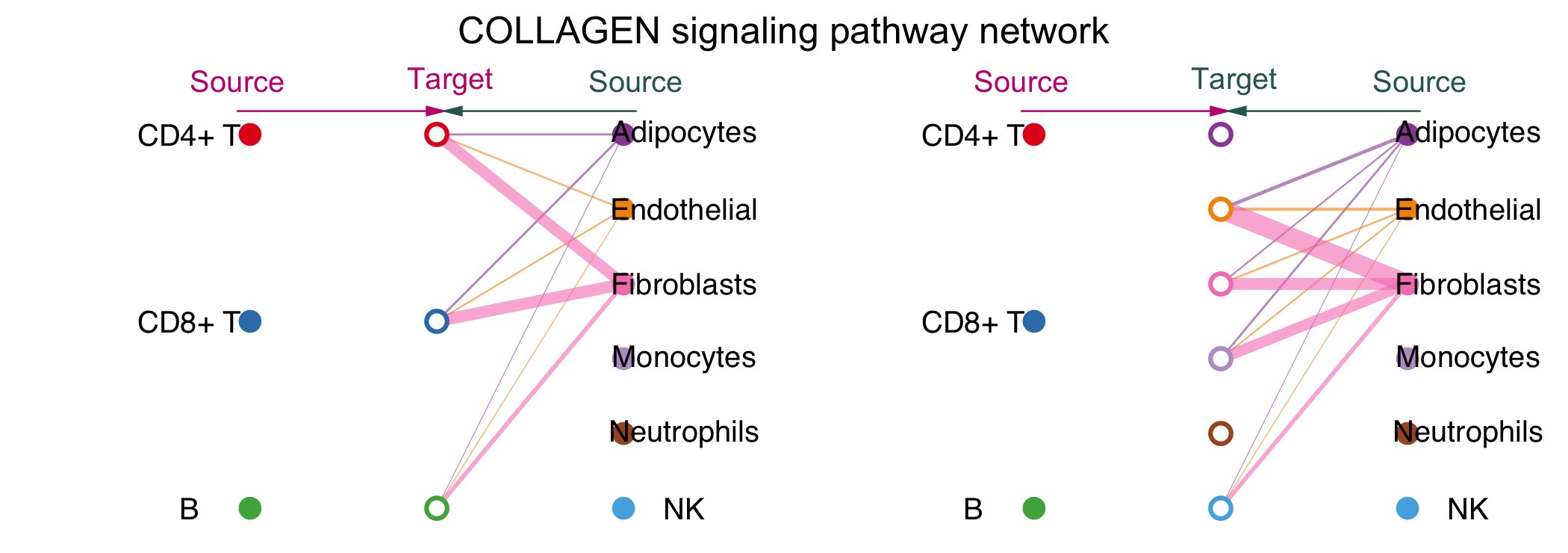
Circle图
不同颜色代表不同细胞类型,结合箭头代表作用方向,粗细代表互作的强度
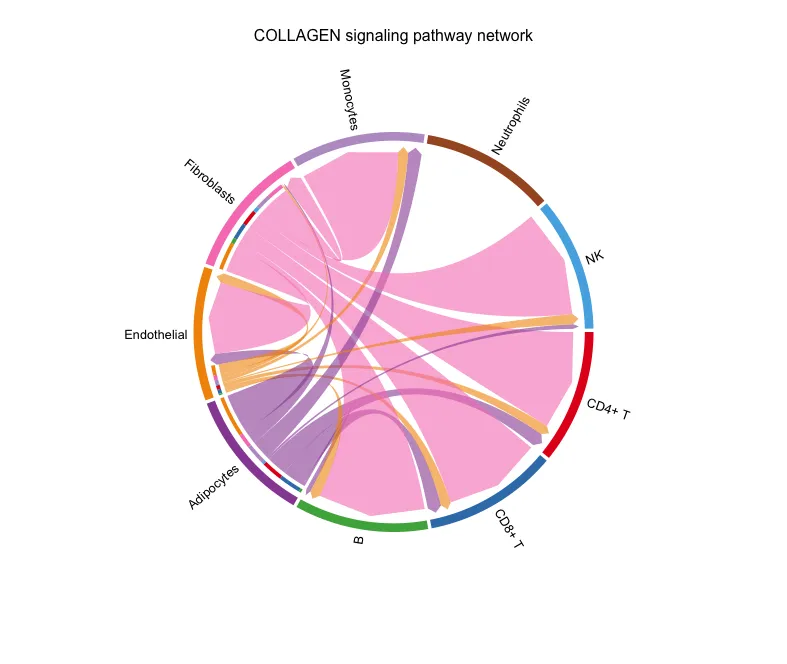
弦图
图片的解读跟上面的图类似。
分组弦图
对T和B统一设置成了T/B,因此图形中属于T/B组的细胞的颜色就变成了一样(这里是粉色)
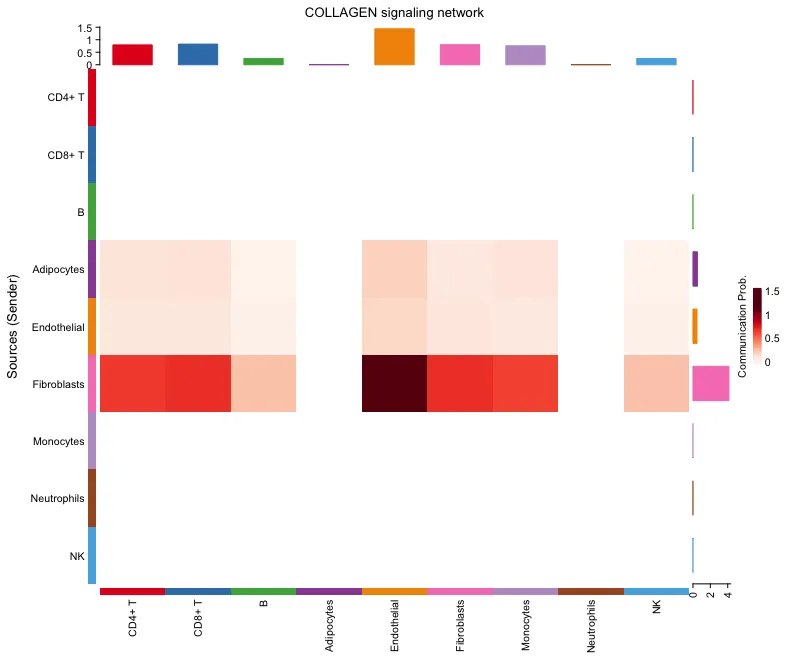
热图
行(Sources - 发送方): 热图的行代表了不同的细胞类型,这些细胞作为信号的发送者(Source)。
列(Targets - 接收方): 热图的列代表了不同的细胞类型,这些细胞作为信号的接收者(Target)。
颜色深浅: 热图中的颜色深浅表示了通讯概率的大小。颜色越深,表示通讯概率越高,这意味着发送方细胞和接收方细胞之间的信号传递越强。
通信概率(Communication Prob.): 右侧的颜色条是颜色映射的参考。图中的深红色表示较高的通讯概率(靠近1.5),浅色表示较低的通讯概率(靠近0或更低)。
顶部数值范围 (0 - 1.5): 顶部红框中显示的数值范围是 Communication Probability(通信概率),即不同颜色对应的通信概率。这个数值表示在不同细胞类型之间,某个信号通路的通信概率大小。0表示没有通信,1.5表示相对较高的通信概率。
右侧部分的数值范围 (0 - 4): 右侧红框中显示的数值范围则表示的是通信路径的数量(Number of Communications)。0表示没有通信路径,4表示有多个通信路径被计算出来并显示在热图中。
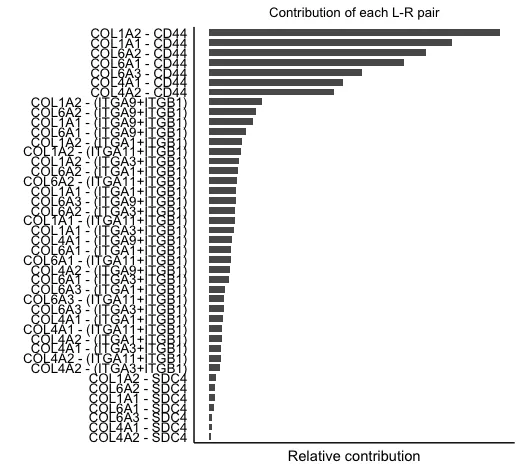
11.计算配-受体对信号通路的贡献并可视化
# 计算配-受体对信号通路的贡献并可视化
netAnalysis_contribution(cellchat, signaling = pathways.show)
# 可视化由单个配体-受体对介导的细胞间通讯
pairLR <- extractEnrichedLR(cellchat, signaling = pathways.show,
geneLR.return = FALSE)
LR.show <- pairLR[1,] # show one ligand-receptor pair
# Hierarchy plot
vertex.receiver = seq(1,3) # a numeric vector
netVisual_individual(cellchat, signaling = pathways.show,
pairLR.use = LR.show,
vertex.receiver = vertex.receiver,
layout = "hierarchy")这里面综合了该数据中在collagen通路中的配-受体贡献。
可视化其中一个配-受体信号情况。
层次结构图:
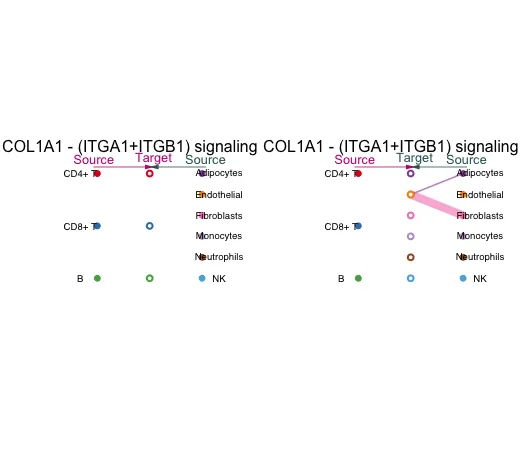
circle图
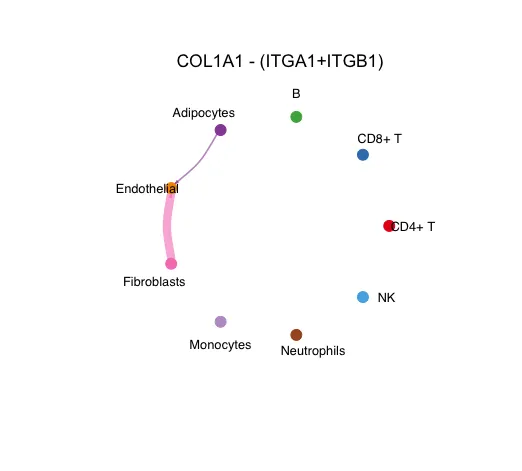
12.官方的存图代码
说实话不好用,还是自己调整之后导图比较好
# Access all the signaling pathways showing significant communications
pathways.show.all <- cellchat@netP$pathways
# check the order of cell identity to set suitable vertex.receiver
levels(cellchat@idents)
vertex.receiver = seq(1,3)
for (i in 1:length(pathways.show.all)) {
# Visualize communication network associated with both signaling pathway and individual L-R pairs
netVisual(cellchat, signaling = pathways.show.all[i], vertex.receiver = vertex.receiver, layout = "hierarchy")
# Compute and visualize the contribution of each ligand-receptor pair to the overall signaling pathway
gg <- netAnalysis_contribution(cellchat, signaling = pathways.show.all[i])
ggsave(filename=paste0(pathways.show.all[i], "_L-R_contribution.pdf"), plot=gg, width = 3, height = 2, units = 'in', dpi = 300)
}
系统分析细胞间通讯网络:
为了更好地理解复杂的细胞间通信网络,CellChat 使用了一些来自不同领域的数学和计算方法来定量地测量和分析这些网络。这些方法包括图论(graph theory)、模式识别(pattern recognition)和流形学习(manifold learning)。
上面三种方法的定义:
● Graph Theory(图论):用于构建和分析细胞间通信网络的结构属性,如连接性、中心性等。
● Pattern Recognition(模式识别):用于从通信数据中自动识别和分类特定的通信模式,帮助发现特定配体-受体对的功能角色。
● Manifold Learning(流形学习):用于高维通信数据的降维和可视化,帮助揭示复杂的非线性通信关系
-
CellChat可以利用网络分析中的中心性指标,确定在特定信号网络中,哪些细胞是主要的信号来源、目标、媒介者以及影响者。
-
它可以通过模式识别方法,预测特定细胞类型的关键输入和输出信号,以及不同细胞类型之间的协调反应。
-
CellChat还能通过定义相似性度量,并从功能和拓扑结构的角度进行流形学习,将信号通路进行分组。
-
它还可以通过对多个网络进行联合流形学习,来区分保守的(通用的)和特定情境下的信号通路。
总之CellChat可以通过计算每个细胞群体的多个网络中心性指标,识别出在细胞间通信网络中的主要发送者、接收者、中介者和影响者。具体来说,开发者使用了加权有向网络中的几个指标,包括出度(out-degree)、入度(in-degree)、流量介数(flow betweenness)和信息中心性(information centrality),来分别确定哪些细胞群体是主要的信号发送者、接收者、中介者和影响者。
13.基于配-受体结果进一步可视化
Bubble plot和netVisual_bubble显示一些单元组与其他单元组之间的所有重要相互作用
# 需要指定source和target
# sources.use是发出信号的细胞系,target.use是接受信号的细胞系
levels(cellchat@idents)
netVisual_bubble(cellchat, sources.use = seq(1:3),
targets.use = c(4:10), remove.isolate = FALSE)
ggsave("bubbleplot.pdf",width = 8,height = 16)
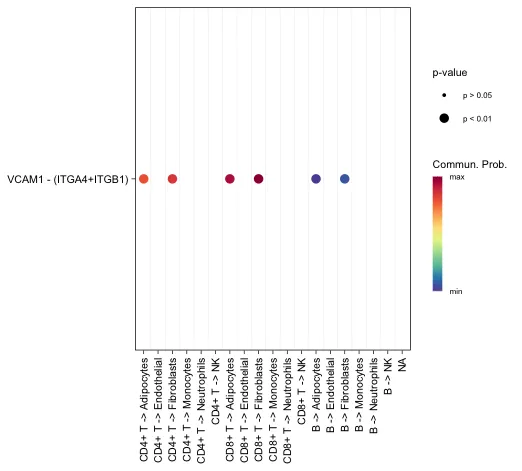
# 还可以增加signaling参数用于展示特定的配受体
cellchat@netP$pathways
netVisual_bubble(cellchat, sources.use = seq(1:3),
targets.use = c(4:10),
signaling = c("VCAM"),
remove.isolate = FALSE)
# 自定义signaling输入展示
# pairLR.use <- extractEnrichedLR(cellchat, signaling = c("CCL","CXCL","FGF"))
# netVisual_bubble(cellchat, sources.use = c(1:3),
# targets.use = c(4:10),
# pairLR.use = pairLR.use,
# remove.isolate = TRUE)
# 可以通过增加下面的参数去设置X轴上的顺序
# sort.by.target = T
# sort.by.source = T
# sort.by.source = T, sort.by.target = T
# sort.by.source = T, sort.by.target = T, sort.by.source.priority = FALSE横轴代表不同细胞之间的互作方向。纵轴代表特定的配-受体对。圆点的不同颜色代表通讯概率。点的大小代表通讯显著性的P值。
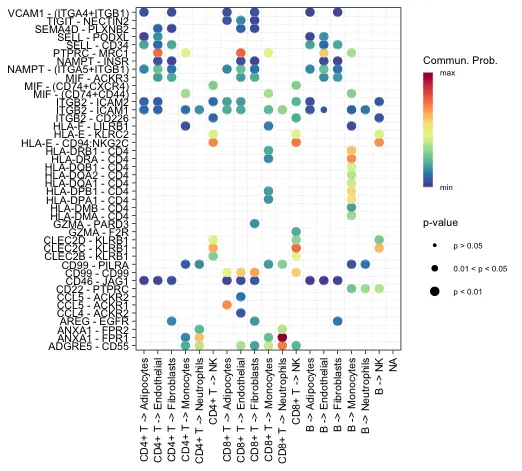
展示特定通路的情况
14. Chord diagram
# 这里进行绘制时建议设定singaling或者减少互作的细胞
# 内容大多会导致不出图
cellchat@netP$pathways
netVisual_chord_gene(cellchat, sources.use = c(1:3),
targets.use = c(4:10),
#signaling = c("COLLAGEN"),
lab.cex = 0.5,
legend.pos.y = 30)
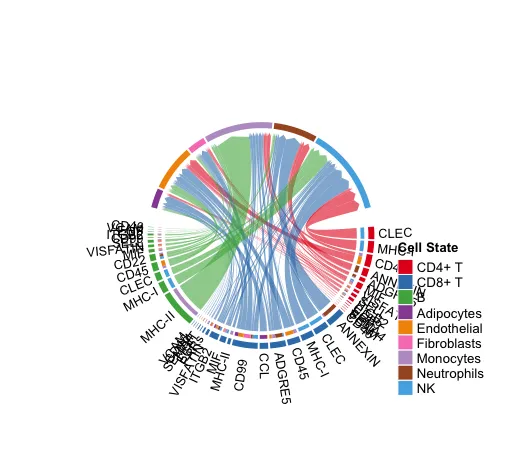
# 显示从某些细胞组(sources.use)到其他细胞组(targets.use)的所有重要信号通路。
netVisual_chord_gene(cellchat, sources.use = c(1:3),
targets.use = c(4:10),
slot.name = "netP",
legend.pos.x = 10)
# NB: Please ignore the note when generating the plot such as "Note: The first link end is drawn out of sector 'MIF'.". If the gene names are overlapped, you can adjust the argument small.gap by decreasing the value.使用者自行分析的时候不要展示太多的配-受体情况,太多就会出现图中的这种情况。
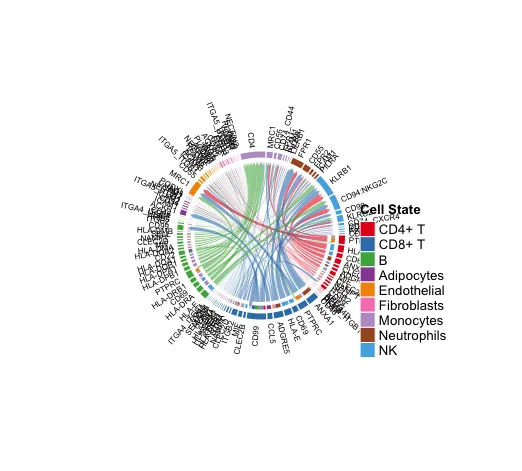
显示从某些细胞组(sources.use)到其他细胞组(targets.use)的所有重要信号通路
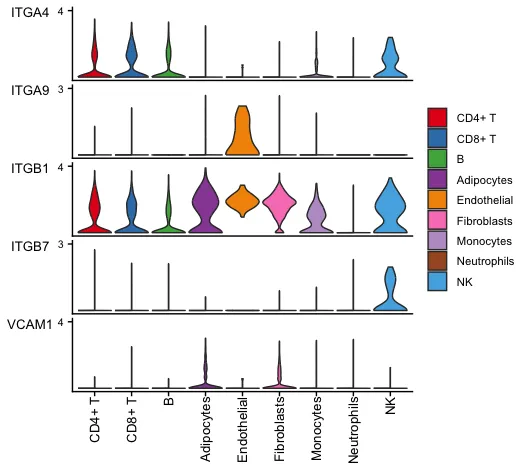
15.使用小提琴/点图绘制信号通路的基因表达情况
# CellChat可以使用Seurat包装函数plotGeneExpression绘制与 L-R 对或信号通路相关的信号转导基因的基因表达分布。
# 该功能提供 "violin"、"dot"、"bar" 三种类型的可视化。
# 或用户可以使用 extractEnrichedLR 提取与推断的 L-R 对或信号通路相关的信号转导基因,然后使用Seurat或其他软件包绘制基因表达。
plotGeneExpression(cellchat, signaling = "VCAM",
enriched.only = TRUE,
type = "violin")显示某一条通路在不同细胞系中的关键基因表达情况
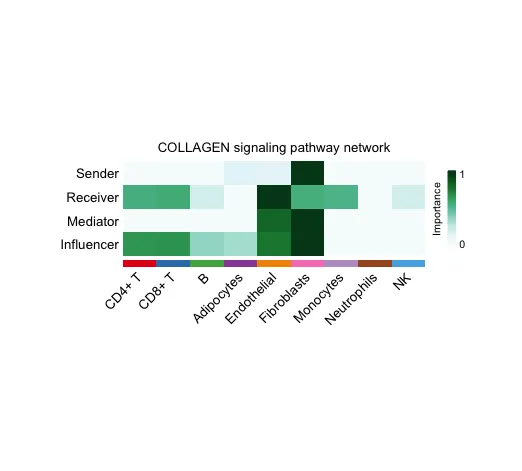
16. 系统分析细胞间通讯网络计算并可视化网络中心性得分
# Compute the network centrality scores
cellchat <- netAnalysis_computeCentrality(cellchat,
slot.name = "netP")
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show,
width = 8, height = 2.5, font.size = 10)行(Sender, Receiver, Mediator, Influencer):行表示在信号通路网络中,不同细胞类型扮演的角色:
● Sender:信号的发送者,即哪些细胞类型是主要的信号发出者。
● Receiver:信号的接收者,即哪些细胞类型是主要的信号接收者。
● Mediator:中介者,即在信号传递过程中起到中介作用的细胞类型。
● Influencer:影响者,即在整个信号通路网络中对通信过程产生显著影响的细胞类型。
列(不同的细胞类型):列表示具体的细胞类型,如 CD4+ T 细胞、B 细胞、成纤维细胞(Fibroblasts)等。
颜色深浅(Importance):颜色的深浅表示每个细胞类型在特定角色中的重要性。颜色越深,表示该细胞类型在这个角色中的重要性越高(例如信号传递的强度或频率越大);颜色越浅,表示该细胞类型在这个角色中的重要性较低。
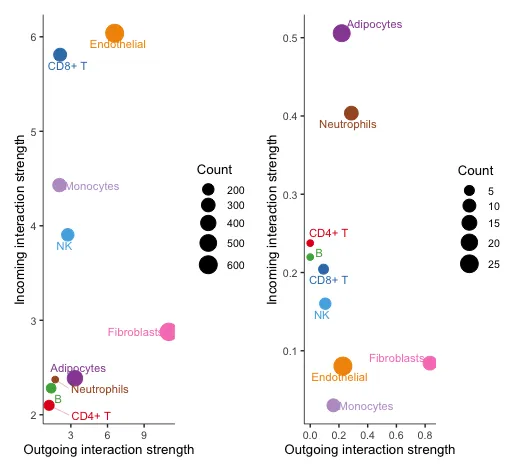
17 在二维空间中可视化主要的sender(Source)和receivers(targets)
# 从所有信号通路对聚合细胞-细胞通信网络的信号作用分析
p1 <- netAnalysis_signalingRole_scatter(cellchat)
# 对特定细胞间通讯网络的信号作用分析
p2 <- netAnalysis_signalingRole_scatter(cellchat, signaling = c("CXCL", "CCL"))
p1 + p2左边代表所有信号的综合评估,可以看到Fibroblasts在输出信号方面的强度很大,Endothelial在接受信号方面的强度很大。
右边代表特定信号的评估,这里选择了CXCL和CCL配受体信号。可以看到还是Fibroblasts在输出信号方面的强度很大,而Adipocytes在接受信号方面的强度很大。
18.识别对某些细胞群的输出或输入信号贡献最大的信号
# pdf("ht1.pdf",width = 12,height = 18)
# ht1 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "outgoing")
# dev.off()
#
# pdf("ht2.pdf",width = 12,height = 18)
# ht2 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "incoming")
# dev.off()
# ht1 + ht2
# class(ht1)
# 特定的signaling
htout <- netAnalysis_signalingRole_heatmap(cellchat,
pattern = "outgoing",
signaling = c("CXCL", "CCL"))
htout
htcome <- netAnalysis_signalingRole_heatmap(cellchat,
pattern = "incoming",
signaling = c("CXCL", "CCL"))
htcome记得筛选一下,不要用全部的,图片会很难看。


除了探索单个信号通路的详细通信外,同时还可以进一步探索多个细胞群体和信号通路如何协调运作。
CellChat 采用了一种模式识别方法来识别这些全局通信模式。 随着模式数量的增加,可能会出现一些的冗余模式,这会使通信模式的解释变得困难。因此,我们默认选择了五种模式进行分析。一般来说,当模式数量超过2时,在生物学上更有意义。
此外,开发者还提供了一个 selectK 函数来推测合适的模式数量。这个函数基于两个已经在 NMF R 包中实现的指标:Cophenetic 和 Silhouette。这两个指标通过对共识矩阵的层次聚类,衡量特定模式数量的稳定性。在一系列模式数量中,当 Cophenetic(Cophenetic 相关系数) 和 Silhouette (轮廓系数)值开始突然下降时,这通常表示适合的模式数量。(Cophenetic 值越接近 1,表示聚类树能够较好地保持原始数据点之间的距离关系,聚类的结果越好, 如果 Cophenetic 值较低,说明聚类树可能失真,不能很好地反映原始数据结构; Silhouette 值用于衡量单个数据点在聚类中的合理性。它评估的是数据点与其所在的簇内点的距离与其离开最近的其他簇的距离之间的比较, Silhouette 值为越是靠近1越能表示数据点很好地匹配了它所在的簇,且与最近的其他簇有明显的差异)。

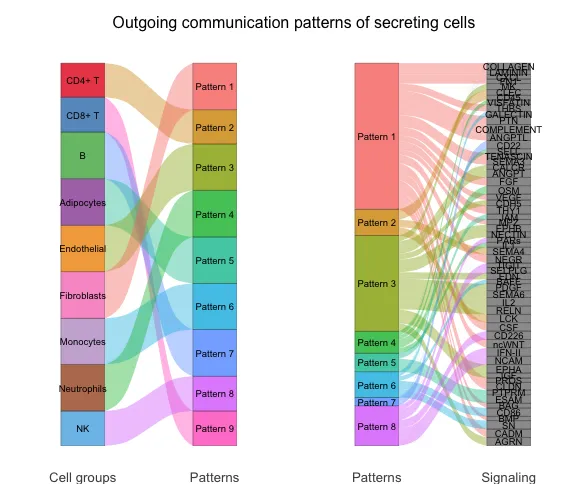
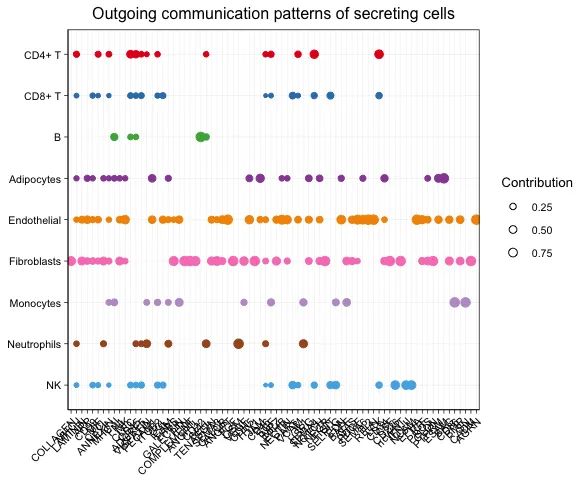
发送模式揭示了发送信号的细胞(即信号来源细胞)如何彼此协调,以及它们如何与某些信号通路协同驱动通信。而接收模式则显示了目标细胞(即信号接收细胞)如何彼此协调,以及它们如何与某些信号通路协同响应外来的信号。
对于发送(或接收)模式,由矩阵分解得到的发送(或接收)细胞间通信概率矩阵中的细胞群体模式矩阵 W 表示这些细胞群体如何协调发送(或接收)信号,而信号通路模式矩阵 H 表示这些信号通路如何协同工作以发送(或接收)信号。为了直观地显示隐含模式与细胞群体以及配体-受体对或信号通路的关联,开发者使用了river图。首先将 W 的每一行和 H 的每一列归一化到 0,1 范围内,然后将 W 和 H 中小于阈值(默认0.5)的元素设置为零。这样的阈值处理可以揭示与每个推断模式相关的最富集的细胞群体和信号通路。经过阈值处理的矩阵 W 和 H 被用作创建river图的输入。
此外,为了直接关联细胞群体与其富集的信号通路,开发者将 W 和 H 中小于阈值的元素设置为零(默认阈值:1/R,其中 R 是潜在模式的数量)。通过使用较不严格的阈值,可以获得与每个细胞群体相关的更多富集信号通路。通过将 W 与 H 相乘计算每个细胞群体对每个信号通路的贡献分数,开发者构建了一个点图,点的大小与贡献分数成比例,以显示细胞群体与其富集的信号通路之间的关联。用户还可以降低参数阈值以显示与每个细胞群体相关的更多富集信号通路。
笔者对这块内容的理解是开发者除了想要探索不同细胞系之间的配-受体/通路互作信息,团队还希望把不同细胞跟整体的配-受体/通路互作信息联系在一起(由"局部"往"整体"拓展),因此通过了非负矩阵分解去识别整体中包含多少"重要"特征,并把这些特征与不同细胞系的发出/接受信号情况联系在一起。
19.识别整体通信模式/以探索多种细胞类型和信号通路如何协调运作
outgoing
library(NMF)
library(ggalluvial)
selectK(cellchat, pattern = "outgoing")
# 当输出模式的数量为9时,Cophenetic值和Silhouette值都开始突然下降。
nPatterns = 9
cellchat <- identifyCommunicationPatterns(cellchat,
pattern = "outgoing",
k = nPatterns)
# river plot
netAnalysis_river(cellchat, pattern = "outgoing")
# dot plot
netAnalysis_dot(cellchat, pattern = "outgoing")k值分析
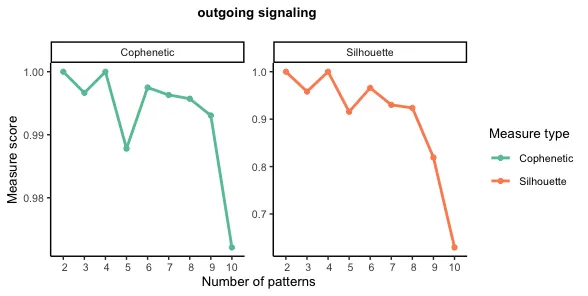
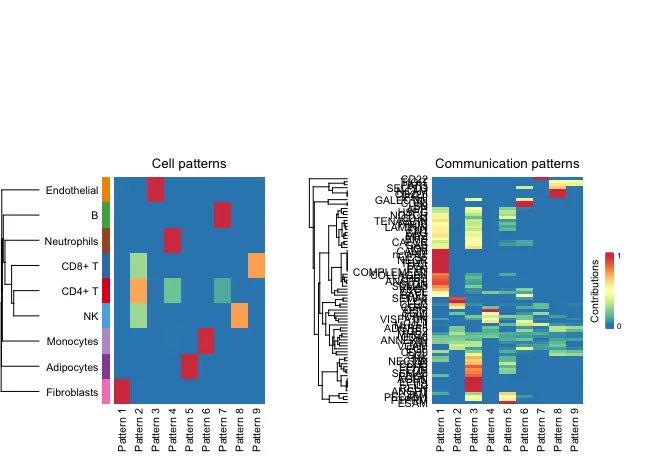
设定特定K值之后的细胞模式(Cell Patterns)和 通讯模式(Communication Patterns) 的关联分析结果。
左图---细胞模式(Cell Patterns) :
树状图(Dendrogram):显示了不同细胞类型之间的相似性或关系。越接近的分支表示这些细胞类型在通讯模式上越相似。
颜色块(Heatmap):表示每个细胞类型在不同模式(Pattern 1 到 Pattern 9)中的贡献或参与度。颜色越深(例如红色),表示该细胞类型在该模式中的贡献越大。
右图---通讯模式(Communication Patterns) :
树状图(Dendrogram):显示了不同通讯模式之间的相似性。
颜色块(Heatmap):表示不同信号通路(如 GAL、COMP、SPP1 等)在不同模式中的贡献。颜色越深(例如红色),表示该信号通路在该模式中的贡献越大
River图
Dot图
incoming
selectK(cellchat, pattern = "incoming")
# 当输出模式的数量为6时,Cophenetic值和Silhouette值都开始突然下降。
nPatterns = 8
cellchat <- identifyCommunicationPatterns(cellchat,
pattern = "incoming",
k = nPatterns)
# river plot
netAnalysis_river(cellchat, pattern = "incoming")
# dot plot
netAnalysis_dot(cellchat, pattern = "incoming")这里就不展示图了。

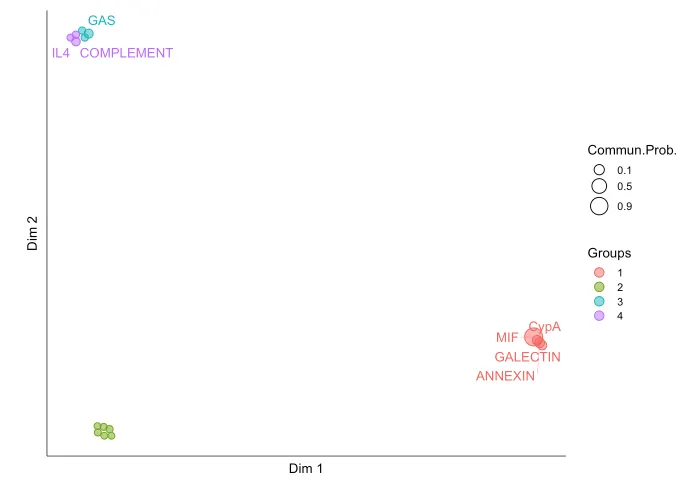
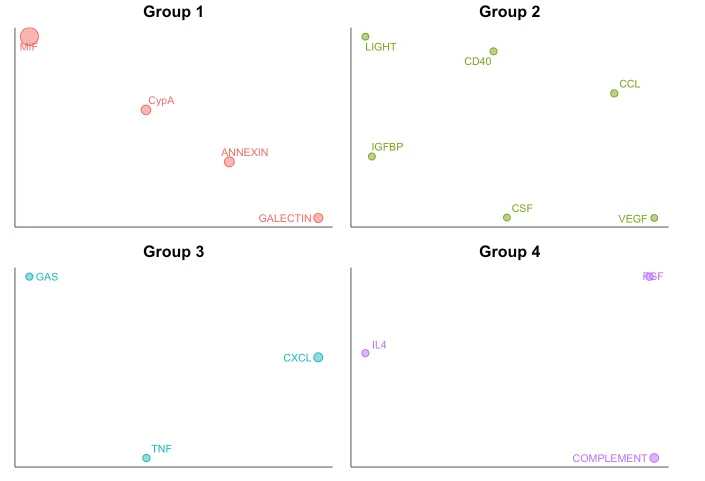
CellChat 能够量化所有重要信号通路之间的相似性,并根据它们在细胞通信网络中的相似性将它们分组。分组情况是基于functional/structural相似性进行展示。
功能相似性分析主要关注信号发送者和接收者之间的相似性。高功能相似性意味着主要的信号发送者和接收者在不同的信号通路之间是相似的。这通常表明这些信号通路在生物学功能上可能具有类似的作用,或者它们的作用是冗余的。
结构相似性分析主要关注信号网络的整体结构,而不考虑发送者和接收者之间的具体相似性。这种分析方法着重于信号网络的拓扑结构,通过比较网络结构本身的相似性来分组。
20.信号网络的流形学习和分类学习分析
cellchat <- computeNetSimilarity(cellchat, type = "functional")
cellchat <- netEmbedding(cellchat, type = "functional")
# 运行clustering
cellchat <- runnetClustering(cellchat,
type = c("functional"),
nCores = 1)
# Visualization in 2D-space
netVisual_embedding(cellchat,
type = c("functional"),
label.size = 3.5)
# 基于结构相似性识别信号组
cellchat <- computeNetSimilarity(cellchat, type = "structural")
cellchat <- netEmbedding(cellchat, type = "structural")
cellchat <- runnetClustering(cellchat, type = "structural",nCores = 1)
# Visualization in 2D-space
netVisual_embedding(cellchat,
type = "structural",
label.size = 3.5)
netVisual_embeddingZoomIn(cellchat,
type = c("structural"),
nCol = 2)会把功能/结构相似的通路分配在一起~
debug:

如果需要这种报错,可以采用两种方式,一种是运行"cellchat <- updateCellChat(cellchat)"代码,另外一种是把igraph降级到1.3.5版本
参考资料:
1、Identify signaling groups based on structure similarityInference and analysis of cell-cell communication using CellChat . Nat Commun. 2021 Feb 17;12(1):1088.
2、cellchat V2 :
https://github.com/jinworks/CellChat?tab=readme-ov-file
3、单细胞天地:
https://mp.weixin.qq.com/s/Pz_pEv4RooGKKzNBphWnRw
4、生信菜鸟团:
https://mp.weixin.qq.com/s/g4j7u0gHmpJH9ZA1Jp9hCw
https://mp.weixin.qq.com/s/npOyrOufgrZtivVFcvtlkQ
5、生信技能树:
https://mp.weixin.qq.com/s/9Q4rxGKFo_jD8vBkuOzTbQ
6、YuLabSMU:
https://mp.weixin.qq.com/s/wQJzTxf-hKUB_jIqLGSeZg
7、生信随笔:
https://mp.weixin.qq.com/s/Tw6FH2vqLghfIQ7kxUthWw
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。
- END -