
引言
今天给大家介绍的是基于 Sentieon 软件开发的用于小麦全基因组测序数据的自动化流程脚本。该流程实现了从原始测序数据(FASTQ)到变异检测结果(GVCF)以及joint calling的完整分析流程,支持多个测序平台和输出格式。
脚本支持原始测序数据(raw_fastq)、过滤后的测序数据(clean_fastq),进行质控、比对、排序、标记重复、生成质量评估指标,最终通过Haplotyper 算法进行变异检测,输出 gVCF 文件。
测试小麦样本平均测序深度7.55x,从FastQ到VCF全流程分析最快用时1.4个小时,大幅缩短小麦全基因组WGS分析时间,有效加快小麦的分子育种进程。
感谢Ampere Computing LLC 和比亚迪服务器部门对本次测试的大力支持!!!
1. 环境设置与参数解析
1.1 Sentieon环境配置
1.1.1 下载地址
软件地址链接
模型下载链接(请转到VX查看链接)
1.1.2 环境设置变量
要开始使用Sentieon®软件,您需要以下条件:
硬件要求
具有以下配置的Linux服务器:
- 运行以下或更高版本的Linux发行版之一:RedHat/CentOS 6.5、Debian 7.7、 OpenSUSE-13.2或Ubuntu-14.04。
- 对于小型面板或全外显子组测序,需16GB内存;对于全基因组测序,需64GB内存。
- (推荐)最好使用高速SSD驱动器,以获得理想的I/O性能,实现最大CPU利用率。
软件要求
- 需要Python 2.6.x、Python 2.7.x或python3.x。您可以通过以下命令检查Python版本:
css
python --version软件安装包:
-
下载软件安装包(以v202308.03版本为例,更新版本需要联系sentieon@insvast.com):
-
使用以下命令解压软件包,其中VERSION为您使用的版本,例如202308.03:
tar xvzf sentieon-genomics-VERSION.tar.gz
许可证要求
Sentieon®软件是一个商用许可的软件。用户需要正确设置许可证才能运行软件。我们提供两种类型的许可证:
- 单机评估许可证:此许可证用于在单台机器上评估Sentieon®软件。它允许新用户快速开始使用软件,而无需IT部门的帮助。要使用此许可证,计划运行Sentieon®软件的计算机需要外部Internet访问。
- 集群许可证:此许可证用于集群环境。使用此许可证,一个轻量级的浮动许可证服务器进程在集群中的一个节点上运行,通过TCP向所有其他与许可证服务器有网络连接的节点提供许可证。此许可证服务器在集群外围的一个特殊非计算节点上运行,该节点可以通过HTTPS不受限制地访问外部世界,并通过监听集群内需要开放的特定TCP端口向集群中的其余节点提供许可证。
(1)设置单机评估许可证
要使用单机评估许可证,计算节点需要能够访问Internet。这允许Sentieon®软件验证许可证。
要使用单机评估许可证,请按照以下步骤操作:
- 将许可证文件复制到计算节点。例如,许可证文件LICENSE_FILE.lic现在位于LICENSE_DIR。
- 按如下方式设置环境变量:
ini
export SENTIEON_LICENSE=LICENSE_DIR/LICENSE_FILE.lic(2)设置许可证服务器
如下面Sentieon软件授权许可拓扑图,许可证服务器需要满足以下条件:
- 许可证服务器应该能够访问Internet以执行许可证验证。
- 计算节点应该能够通过主机名LICSRVR_HOST访问许可证服务器。
- 运行许可证服务器的机器有一个开放的端口供许可证服务监听,并且计算节点可以访问该端口。这里我们假设可用端口是LICSRVR_PORT。
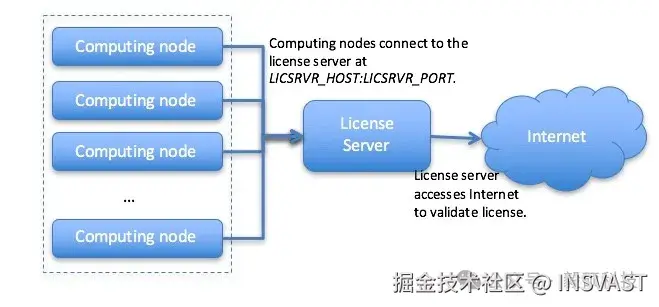
您可能需要IT同事支持来获取LICSRVR_HOST:LICSRVR_PORT,并确认满足上述要求。
注意:如果许可证服务器位于防火墙后面,通过NAT与计算节点分离,则节点可见的许可证服务器主机名/IP可能与其实际主机名/IP不同。如果是这种情况,您需要将许可证服务器绑定到实际IP地址,而计算节点从NAT后的IP地址请求许可证。请联系sentieon@insvast.com以获取更多详细信息。
按照以下步骤获取许可证文件,设置和测试许可证服务器:
1)将以下信息发送给sentieon@insvast.com以接收许可证文件:
- 指定运行许可证服务的机器的FQDN(Hostname)LICSRVR_HOST。
- 指定的端口LICSRVR_PORT。
2)将收到的许可证文件复制到许可证服务器LICSRVR_HOST。我们假设许可证文件位于LICENSE_PATH/LICENSE_FILE。在许可证服务器上运行以下命令以启动许可证服务器进程:
bash
<SENTIEON_INSTALL_DIR>/bin/sentieon licsrvr --start --log LOG_FILE LICENSE_PATH/LICENSE_FILE3)或者,您可以按照以下的说明,将许可证服务器配置并启动为系统守护进程。
使用sysvint将许可证服务器作为系统服务运行
如果您的系统遵循传统的System V init启动脚本,您可以通过运行以下命令作为root用户来设置许可证服务器,使其在系统启动时自动启动:
- 创建并自定义配置文件:配置文件通常为/etc/sysconfig/licsrvr;但在Ubuntu中,配置文件将是/etc/default/licsrvr。以下是配置文件的示例,推荐使用以下设置:
arduino
/home/sentieon/release/latest 是最新Sentieon®软件包安装目录的符号链接
/home/sentieon/licsrvr 是运行licsrvr服务的文件夹
/home/sentieon/licsrvr/licsrvr.lic 是Sentieon®许可证文件
licsrvr="/home/sentieon/release/latest/bin/sentieon
licsrvr"licfile="/home/sentieon/licsrvr/licsrvr.lic"
logfile="/home/sentieon/licsrvr/licsrvr.log"- 将许可证服务器启动脚本安装到/etc/init.d目录。启动脚本包含在发布包中。
bash
install -m 0755 $SENTIEON_INSTALL_DIR/doc/licsrvr.sh /etc/init.d/licsrvr-
安装并启用服务。根据您的系统,您将运行不同的命令:
-
如果您的系统安装了Linux Standard Base Core Specifications,执行系统init脚本安装脚本:
bash
/usr/lib/lsb/install_initd /etc/init.d/licsrvr- 如果您的系统没有安装lsb.conformance包,使用chkconfig命令启用服务:
csharp
chkconfig --add licsrvrchkconfig licsrvr on- 对于Ubuntu和Debian系统,如果您没有lsb/install_initd二进制文件且选择不安装lsb-core包,请使用update-rc.d命令安装并启用服务:
bash
update-rc.d licsrvr defaultsupdate-rc.d licsrvr enable- 您可以使用service命令来启动/停止/重启/检查服务的状态:
arduino
service licsrvr {start|stop|restart|status}使用systemd将许可证服务器作为系统服务运行
您可以使用操作系统的systemd系统和服务功能来设置许可证服务器,使其在系统中自动启动。要做到这一点,请以root用户身份运行以下命令:
- 如果您使用doc文件夹中的licsrvr.service许可证服务器启动脚本,您需要创建脚本所需的必要文件,包括使用sentieon用户名:
- /home/sentieon/release/latest 是最新Sentieon®软件包安装目录的符号链接
- /home/sentieon/licsrvr 是运行licsrvr服务的文件夹
- /home/sentieon/licsrvr/licsrvr.lic 是Sentieon®许可证文件
或者,您可以编辑许可证服务器启动脚本以指向您的特定用户名和/或位置信息。
- 将许可证服务器启动脚本安装到/etc/systemd/system目录:
bash
install -m 0644 $SENTIEON_INSTALL_DIR/doc/licsrvr.service /etc/systemd/system- 运行以下命令以启用许可证服务器在计算机启动时自动启动:
bash
systemctl enable licsrvr.service- 您可以使用systemctl命令手动启动和停止服务:
arduino
systemctl start licsrvr.servicesystemctl stop licsrvr.service- 进入Sentieon®安装目录。在许可证服务器上运行以下命令,确认许可证服务器已启动并正在运行。
javascript
<SENTIEON_INSTALL_DIR>/bin/sentieon licclnt ping -s LICSRVR_HOST:LICSRVR_PORT如果命令返回时没有错误消息,则许可证服务器已启动并正在运行。
- 登录到其中一个计算节点,进入Sentieon®安装目录,再次运行上述命令:
javascript
<SENTIEON_INSTALL_DIR>/bin/sentieon licclnt ping -s LICSRVR_HOST:LICSRVR_PORT如果命令返回时没有错误消息,则计算节点现在也可以访问许可证服务器。
- 设置以下环境变量,您就可以开始使用了。
ini
export SENTIEON_LICENSE=LICSRVR_HOST:LICSRVR_PORT系统环境要求
- 如果Python 2.6.x、Python 2.7.x或python3.x不是默认Python版本,您可以设置以下环境变量:
ini
export SENTIEON_PYTHON=Python_location- 如果使用本地主机许可证文件,请设置以下环境变量,其中LICENSE_DIR为许可证文件所在目录,LICENSE_FILE.lic为许可证文件名:
ini
export SENTIEON_LICENSE=LICENSE_DIR/LICENSE_FILE.lic- 如果用户使用许可证服务器,请设置以下环境变量,其中LICSRVR_HOST和LICSRVR_PORT分别为许可证服务器的主机名和端口。详情请参阅下一节。
ini
export SENTIEON_LICENSE=LICSRVR_HOST:LICSRVR_PORT- 为方便起见,请按如下方式设置二进制路径,其中PATH_TO_SENTIEON_BINARY_DIRECTORY为Sentieon®二进制文件的安装目录:
ini
export SENTIEON_INSTALL_DIR=PATH_TO_SENTIEON_BINARY_DIRECTORY- 使用NFS存储时,为提高性能,请将SENTIEON_TMPDIR环境变量设置为指向本地快速临时存储:
bash
export SENTIEON_TMPDIR=/tmp1.2 变量定义与参数解析
bash
#!/bin/bash
echo $0 $SAMPLEID $WORKDIR $FASTQ_1 $FASTQ_2 $FASTA $SUFFIX $DATATYPE $KEEP_CLEAN $KEEP_BAM $PLOIDY
set -euxo pipefailecho:打印脚本名及输入参数占位符,方便调试时确认参数顺序。
set -euxo pipefail:设置脚本执行规则,增强健壮性。
ini
export SAMPLEID=$1
export WORKDIR=$2
export FASTQ_1=$3
export FASTQ_2=$4
export FASTA=$5
BSUFFIX=$6
TYPE=${7:-"raw"}
KEEP_CLEAN=${8:-keep}
KEEP_BAM=${9:-keep}
PLOIDY=${10:-2}
LOGFILE=$SAMPLEID.run.log
export root=/Path/sentieon/202503/sentieon-genomics-202503/bin/从命令行读取参数,包括样本 ID、工作目录、输入文件路径等。
部分参数设置默认值。
LOGFILE:定义日志文件名(样本 ID+.run.log)。
root:sentieon工具(基因组分析软件)的安装路径。
更详细的参数表格,如下表所示:
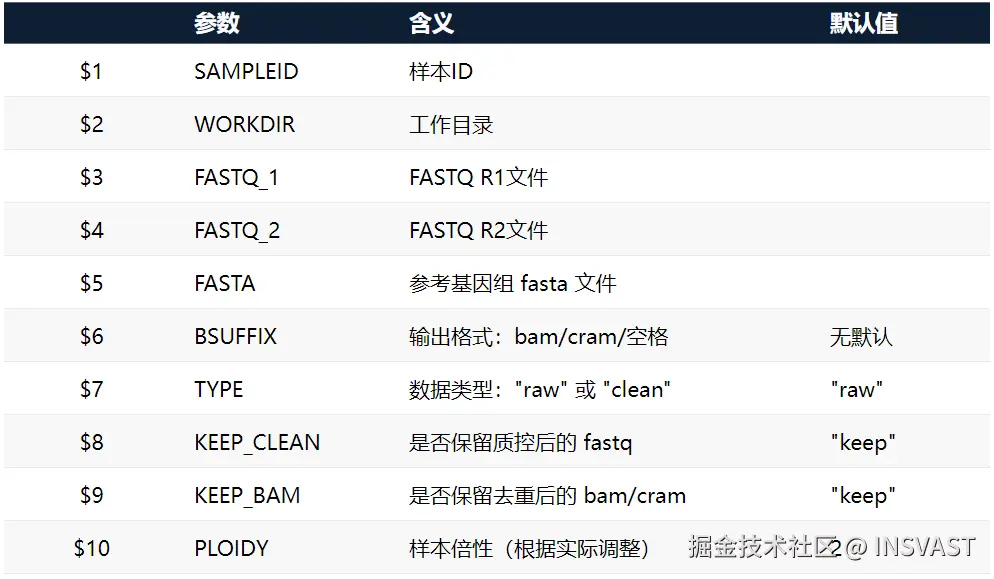
1.3 输入文件有效性检查
bash
if [ "$BSUFFIX" = "bam" ] || [ "$BSUFFIX" = "cram" ] || [ "$BSUFFIX" = " " ]; then
echo "$BSUFFIX check"
else
die "Error: check 6th blank, BSUFFIX must be 'bam' or 'cram' or space"
fi检查BSUFFIX,只能是bam(二进制比对格式)、cram(压缩比对格式)或空格,否则报错退出。
1.4 测序平台判断
ini
if [ -e $LOGFILE ];then
export PLATFORM=$(awk '/Platform/{print $NF;exit}'$LOGFILE)
else
export count=$(zcat $FASTQ_1|head -n 1|awk '{print NF}')
if [ $count -eq 1 ];then
export PLATFORM="DNBSEQ"
elif [ $count == 2 ];then
export PLATFORM="ILLUMINA"
elif [ $count == 3 ];then
export PLATFORM="ILLUMINA"
else
echo"Unrecognized platform"
export PLATFORM="ILLUMINA"
fi
fi若日志文件已存在,从日志中提取测序平台(Platform字段)。
若日志不存在,通过 FASTQ 文件首行的字段数判断平台。
1.5 输出文件格式确定
swift
FAI=$FASTA.fai
if [ "$BSUFFIX" = "bam" ] || [ "$BSUFFIX" = "cram" ];then
export SUFFIX=$(awk -v preset=$BSUFFIX 'BEGIN{max=0}{if($2>max)max=$2}END{if(max>536870911){print "cram"}else{print preset}}' $FAI)
else
export SUFFIX=$(awk 'BEGIN{max=0}{if($2>max)max=$2}END{if(max>536870911){print "cram"}else{print "bam"}}' $FAI)
fi根据参考基因组索引文件($FASTA.fai)中最长序列的长度决定输出比对文件格式:
- 若最长序列长度 > 536870911(约 512MB),强制使用cram(更适合大基因组压缩)。
- 否则根据BSUFFIX或默认bam。
补充说明:对于单个染色体长度>536870911(约512MB)的物种(比如小麦),Sentieon软件可以切换至cram,不用因BAM 文件索引 (.bai) 的格式限制切割染色体。
1.6 环境与目录配置
bash
export TMPDIR=$WORKDIR
export THREADS=$(nproc)
[ -e $WORKDIR ]||mkdir -p $WORKDIR
cd $WORKDIR
exec >>$LOGFILE 2>&1
echo "SampleID:" $SAMPLEID
echo "DataType:" $TYPE
......配置临时目录、线程数,确保工作目录存在,并将所有输出写入日志文件。
1.7 设置计时函数(可选)
bash
timer(){
start_time=$(date +%s)
eval $2 && touch $3
end_time=$(date +%s)
cost_time=$[ $end_time-$start_time ]
echo -e "TIMER: $1\t$(($cost_time/60)) min $(($cost_time%60)) s"
}用于记录每个分析步骤的开始/结束时间、耗时,并通过创建标记文件(如qc.ok)标记步骤完成,避免重复执行。
2. 数据质控(Raw2clean)
bash
raw2clean(){
cmd="fastp -w 16 -i $FASTQ_1 -I $FASTQ_2 -o $clean1 -O $clean2 -j $SAMPLEID.qc.json -h $SAMPLEID.qc.html&&rm $FASTQ_1 $FASTQ_2"
timer raw2clean "$cmd" qc.ok
}使用fastp工具对原始 FASTQ 数据进行质控(过滤低质量reads、接头等),输出过滤后的 FASTQ 文件及质控报告文件(JSON/HTML)。
成功后创建qc.ok标记文件。
3. 序列比对与排序(Alignment)
- 双端测序比对脚本:
bash
alignment(){
tag="@RG\tID:rg_$SAMPLEID\tSM:$SAMPLEID\tPL:$PLATFORM"
cmd="sentieon bwa mem -R "$tag" -t $THREADS -K 10000000 -x $ML_MODEL/bwa.model $FASTA $clean1 $clean2|sentieon util sort --temp_dir $TMPDIR -r $FASTA -o $SAMPLEID.sorted.$SUFFIX -t $THREADS --sam2bam -i -"
echo $cmd
timer alignment "$cmd" align.ok
}- 单端测序比对脚本:
bash
alignment(){
tag="@RG\tID:rg_$SAMPLEID\tSM:$SAMPLEID\tPL:$PLATFORM"
cmd="$root/sentieon bwa mem -R "$tag" -t $THREADS -K 10000000 $FASTA -x $ML_MODEL/bwa.model $clean1 |$root/sentieon util sort --temp_dir $TMPDIR -r $FASTA -o $SAMPLEID.sorted.$SUFFIX -t $THREADS --sam2bam -i -"
echo $cmd
timer alignment "$cmd" align.ok
}使用sentieon bwa turbo进行序列比对(基于BWA算法),添加Read Group信息(用于后续变异分析),并通过sentieon util sort对结果排序,输出sorted.bam或sorted.cram。
基于测序平台选择不同的机器学习模型($ML_MODEL)优化比对。
成功后创建align.ok标记文件。
4. 生成质量评估指标(Metrics)
bash
metrics(){
cmd="sentieon driver --temp_dir $TMPDIR -r $FASTA -t $THREADS -i $SAMPLEID.sorted.$SUFFIX --algo WgsMetricsAlgo $SAMPLEID.WGS_METRICS.txt --algo MeanQualityByCycle $SAMPLEID.mq_metrics.txt --algo QualDistribution $SAMPLEID.qd_metrics.txt --algo GCBias --summary $SAMPLEID.gc_summary.txt $SAMPLEID.gc_metrics.txt --algo AlignmentStat $SAMPLEID.aln_metrics.txt --algo BaseDistributionByCycle $SAMPLEID.bd_metrics.txt --algo QualityYield $SAMPLEID.qy_metrics.txt --algo InsertSizeMetricAlgo $SAMPLEID.is_metrics.txt"
timer metrics "$cmd" metrics.ok
}使用sentieon driver计算多种测序质量指标,包括:
- 全基因组测序指标(WgsMetricsAlgo)。
- 碱基质量分布、GC 偏差、插入片段长度等。
结果输出到多个质量指标的.txt文件,成功后创建metrics.ok。
5. 标记重复序列(Dedup)
bash
dedup(){
cmd="sentieon driver -r $FASTA --temp_dir $TMPDIR -t $THREADS -i $SAMPLEID.sorted.$SUFFIX --algo LocusCollector --fun score_info $SAMPLEID.score.txt&&sentieon driver -r $FASTA -t $THREADS -i $SAMPLEID.sorted.$SUFFIX --algo Dedup --score_info $SAMPLEID.score.txt --metrics $SAMPLEID.dedup_metrics.txt $SAMPLEID.deduped.$SUFFIX&&rm $SAMPLEID.sorted.$SUFFIX* $SAMPLEID.score.txt*"
timer markdup "$cmd" markdup.ok
}标记重复序列分两步:
LocusCollector:收集位点质量分数信息。Dedup:基于分数标记并去除 PCR 重复序列。
完成后删除中间文件(排序后的比对文件),创建markdup.ok。
6. 碱基质量重校正(BQSR可选)
bash
bqsr(){
sentieon driver -t $THREADS -r $FASTA -i $SAMPLEID.deduped.$SUFFIX --algo QualCal -k $KNOWN_SITES $SAMPLEID.RECAL_DATA.TABLE
sentieon driver -t $THREADS -r $FASTA -i $SAMPLEID.deduped.$SUFFIX -q $SAMPLEID.RECAL_DATA.TABLE --algo QualCal -k $KNOWN_SITES $SAMPLEID.RECAL_DATA.TABLE.POST --algo ReadWriter $SAMPLEID.deduped.RECALIBRATED.$SUFFIX
timer markdup "$cmd" markdup.ok
}- 此过程分为两个主要部分:首先创建一个校正模型(生成 .TABLE 文件),然后应用该模型生成报告和碱基质量重校正后的bam。
--algo QualCal: 指定运行的算法是 QualCal(质量校正算法)。KNOWN_SITES:已知变异数据库的VCF文件路径。RECAL_DATA.TABLE:重校准表的位置和文件名。RECAL_DATA.TABLE.POST: 临时性的后重校准表的位置和文件名。--algo ReadWriter:这一步为可选的。Sentieon® 变异检测可以在运行时使用校正前的 BAM 加上重校准表来即时执行重校正。便可以不输出巨大的BAM文件,节省磁盘的空间。- 若选择执行这一步,第七部分变异检测中请将
$SAMPLEID.deduped.RECALIBRATED.$SUFFIX设为输入文件。
7. 变异检测(DNAseq)
perl
dnaseq(){
cmd="sentieon driver --temp_dir $TMPDIR -r $FASTA -t $THREADS -i $SAMPLEID.deduped.$SUFFIX --algo Haplotyper --emit_conf=30 --call_conf=30 --emit_mode gvcf --ploidy $PLOIDY $SAMPLEID.hc.gvcf.gz && md5sum $SAMPLEID.hc.gvcf.gz > $SAMPLEID.hc.gvcf.gz.md5"
timer Haplotyper "$cmd" hc.ok
}使用Haplotyper算法进行单倍型分析,生成 GVCF 文件。
计算 GVCF 文件的 MD5 ,确保文件完整性,成功后创建hc.ok。
8. Joint Calling
8.1 参数检查与使用说明
bash
#!/bin/bash
[ $# -eq 0 ]&&echo Usage: $(basename $0) $FASTA $GVCF_LIST $NUM $DATADIR&&exit
start_time=$(date +%s)如果脚本没有传入任何参数,则打印使用说明并退出。
使用方式:脚本名 FASTA文件 GVCF列表文件 NUM 数据目录
记录脚本开始执行的时间
8.2 设置错误处理
arduino
set -euo pipefail-e: 任何命令失败则退出脚本。
-u: 使用未定义的变量时报错。
-o pipefail: 管道中任何一个命令失败则整个管道失败。
8.3 参数赋值
ini
DATADIR=$4
FASTA=$1 #"$FASTA_DIR/genomeEN_split.fa"
GVCF_LIST=$2
NUM=$3$1: 参考基因组 FASTA 文件路径。
$2: 包含所有样本 GVCF 文件路径的列表文件。
$3: 用于命名输出文件的数字标识(如批次号)。
$4: 数据目录,用于存放输出文件。
8.4 设置线程数、工作目录和日志文件
bash
NT=$(nproc) #number of threads to use in computation, set to number of cores in the server
WORKDIR="$DATADIR/JointCall-${NUM}"
[ -e $WORKDIR ]||mkdir -p $WORKDIR
#[ -e $file ]&&exit 0
cd $WORKDIR
LOGFILE=$WORKDIR/joint-call${NUM}_run.log
exec >$LOGFILE 2>&1NT: 获取当前系统的 CPU 核心数。
WORKDIR: 根据 NUM 创建唯一的工作目录。
如果目录不存在则创建,并进入该目录。
将所有标准输出和标准错误重定向到日志文件。
8.5 执行联合变异检测
bash
root=/APP/u22/x86_com/sentieon/202503/sentieon-genomics-202503
cat $GVCF_LIST|$root/bin/sentieon driver -r $FASTA --algo GVCFtyper \
$WORKDIR/output${NUM}-jc.vcf.gz - || { echo echo "GVCFtyper failed"; exit 1; }cat $GVCF_LIST: 读取 GVCF 文件列表。
sentieon driver: 调用 Sentieon 驱动程序。
-r $FASTA: 指定参考基因组。
--algo GVCFtyper: 使用 GVCFtyper 算法进行联合变异检测。
output${NUM}-jc.vcf.gz: 输出的压缩 VCF 文件。
如果命令失败,输出错误信息并退出。
8.6 计算并输出运行时间(可选)
perl
end_time=$(date +%s)
cost_time=$[ $end_time-$start_time ]
echo "joint calling time is $(($cost_time/60))min $(($cost_time%60))s"
echo "the Joint-calling done at `date +%H:%M:%S` !!!"计算脚本运行的总时间,并以"分:秒"格式输出。最后输出完成时间。
9. 总结
该脚本实现了DNA测序数据从原始测序数据(FASTQ)到变异检测结果(GVCF)以及joint calling的完整分析流程,支持Illumina/DNBSEQ平台,可配置输出格式(BAM/CRAM)和中间文件保留策略,通过标记文件和日志确保流程可进行追溯,适用于大规模基因组变异检测场景。特点包括:
- 参数化: 通过命令行参数灵活控制输入、输出和流程选项。
- 自动化: 自动检测测序平台和参考基因组以决定最佳分析策略。
- 稳健性: 使用 set -euxo pipefail和标记文件实现错误处理和断点续跑。
- 模块化: 将每个分析步骤封装成函数。
- 高效性: 使用商业优化的sentieon工具替代金标准GATK/BWA,速度更快。
- 可追溯性: 详细的日志记录和 MD5 校验确保结果可重现。
要运行此脚本,需要预先安装好 sentieon、fastp等软件,并准备好对应的模型文件 bundle。
10. 脚本应用示例
使用上述脚本对小麦全基因组测序数据分析的测序结果,具体样本信息如下表所示:
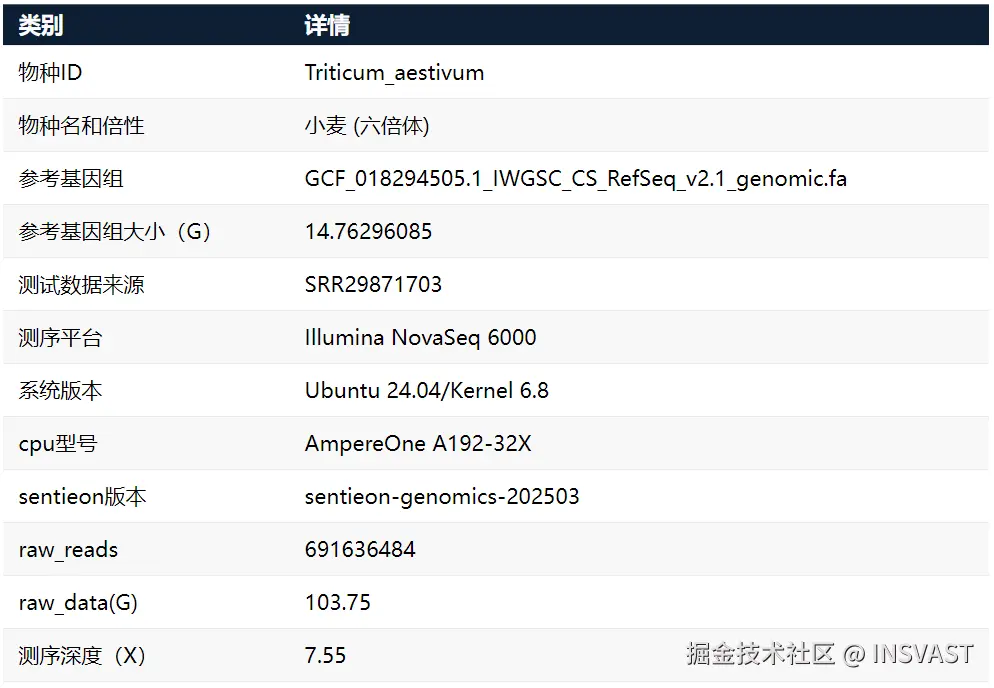
测试样本下载
bash
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/003/SRR29871703/SRR29871703_1.fastq.gz
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/003/SRR29871703/SRR29871703_2.fastq.gz测试硬件配置
- CPU为单颗AmpereOne A192-32X
- 内存为512GB DDR5
- 系统为Ubuntu 24.04/Kernel 6.8
测试结果
使用本文流程对小麦全基因组测序数据进行变异检测分析,下表为不同CPU核数下的计算时间和资源调用情况: 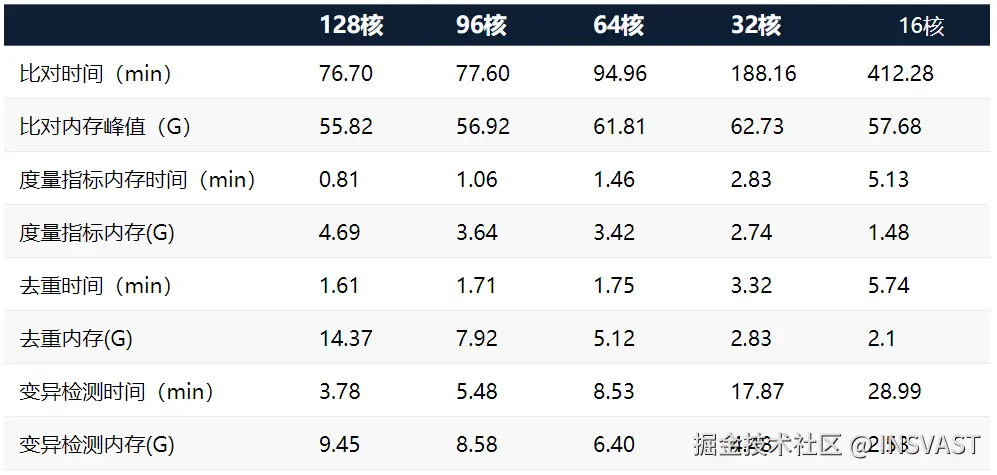
本次测试在不同的线程数上进行性能的比较。从数据中可以明显看出,随着线程数的增加,变异检测的整体效率显著提升。从FastQ到VCF全流程分析最快用时1.4个小时,大幅缩短小麦全基因组WGS分析时间,有效加快小麦的分子育种进程。
Sentieon在不断的优化算法的运行效率,为科研工作者提供更快速、更经济的基因检测方案。若您刚好有需要检测的数据,不妨来申请试用Sentieon吧!
更多下载链接等内容,请关注VX:毅硕科技,了解更多内容。