文章目录
- 一、决策树分类相关理论
-
- [1. 决策树的基本结构](#1. 决策树的基本结构)
- [2. 决策树的分类原理](#2. 决策树的分类原理)
-
- [2.1 树的构建过程(核心阶段)](#2.1 树的构建过程(核心阶段))
- [2.2 样本分类过程](#2.2 样本分类过程)
- [3. 关键核心:特征拆分准则](#3. 关键核心:特征拆分准则)
-
- [3.1 信息增益(Information Gain)与ID3算法](#3.1 信息增益(Information Gain)与ID3算法)
- [3.2 信息增益比(Information Gain Ratio)与C4.5算法](#3.2 信息增益比(Information Gain Ratio)与C4.5算法)
- [3.3 Gini系数与CART算法](#3.3 Gini系数与CART算法)
- [4. 决策树的过拟合与剪枝策略](#4. 决策树的过拟合与剪枝策略)
-
- [4.1 预剪枝(Pre-pruning)](#4.1 预剪枝(Pre-pruning))
- [4.2 后剪枝(Post-pruning)](#4.2 后剪枝(Post-pruning))
- [5. 决策树分类的优缺点](#5. 决策树分类的优缺点)
-
- [5.1 优点](#5.1 优点)
- [5.2 缺点](#5.2 缺点)
- [6. 总结](#6. 总结)
- 二、scikit-learn中决策树分类相关方法介绍
-
- [1. 决策树分类构造方法](#1. 决策树分类构造方法)
-
- [1.1 基础构造语法](#1.1 基础构造语法)
- [1.2 核心构造参数详解](#1.2 核心构造参数详解)
-
- [1.2.1 拆分准则参数](#1.2.1 拆分准则参数)
- [1.2.2 结构控制参数(预剪枝)](#1.2.2 结构控制参数(预剪枝))
- [1.2.3 正则化与其他参数](#1.2.3 正则化与其他参数)
- [1.3 构造前的环境准备](#1.3 构造前的环境准备)
- [2. 决策树分类构造方法返回对象的方法](#2. 决策树分类构造方法返回对象的方法)
-
- [2.1 核心功能方法](#2.1 核心功能方法)
- [2.2 模型可视化与解释方法](#2.2 模型可视化与解释方法)
- [3. 决策树分类构造方法返回对象的属性](#3. 决策树分类构造方法返回对象的属性)
- [4. 决策树分类模型评估指标](#4. 决策树分类模型评估指标)
-
- [4.1 基础评估指标(混淆矩阵衍生指标)](#4.1 基础评估指标(混淆矩阵衍生指标))
- [4.2 综合评估指标](#4.2 综合评估指标)
- [4.3 类别不平衡专用指标](#4.3 类别不平衡专用指标)
- [5. 常见问题与解决方案](#5. 常见问题与解决方案)
-
- [5.1 问题1:模型过拟合(训练集准确率高,测试集准确率低)](#5.1 问题1:模型过拟合(训练集准确率高,测试集准确率低))
- [5.2 问题2:类别不平衡(少数类样本分类准确率低)](#5.2 问题2:类别不平衡(少数类样本分类准确率低))
- [5.3 问题3:可视化失败(plot_tree无输出或export_graphviz报错)](#5.3 问题3:可视化失败(plot_tree无输出或export_graphviz报错))
- [5.4 问题4:模型欠拟合(训练集与测试集准确率均低)](#5.4 问题4:模型欠拟合(训练集与测试集准确率均低))
- 三、基于决策树的威斯康星乳腺癌数据分析与挖掘
-
- [1. 分析目标与技术路线](#1. 分析目标与技术路线)
-
- [1.1 核心目标](#1.1 核心目标)
- [1.2 技术路线](#1.2 技术路线)
- [2. 数据收集与加载](#2. 数据收集与加载)
-
- [2.1 威斯康星乳腺癌数据集介绍](#2.1 威斯康星乳腺癌数据集介绍)
-
- [2.1.1 数据集基本信息](#2.1.1 数据集基本信息)
- [2.1.2 数据集字段说明](#2.1.2 数据集字段说明)
- [2.2 数据加载](#2.2 数据加载)
- [3. 探索性数据分析(EDA)](#3. 探索性数据分析(EDA))
-
- [3.1 数据基本信息](#3.1 数据基本信息)
-
- [3.1.1 查看数据前10行](#3.1.1 查看数据前10行)
- [3.1.2 查看数据摘要信息](#3.1.2 查看数据摘要信息)
- [3.1.3 特征描述性统计](#3.1.3 特征描述性统计)
- [3.2 目标变量类别分布分析](#3.2 目标变量类别分布分析)
- [3.3 目标变量类别与特征相关性分析](#3.3 目标变量类别与特征相关性分析)
- [3.4 关键特征分布分析](#3.4 关键特征分布分析)
- [3.5 关键特征相关性分析](#3.5 关键特征相关性分析)
- [3.6 关键特征的类别区分能力验证](#3.6 关键特征的类别区分能力验证)
- [4. 数据预处理](#4. 数据预处理)
-
- [4.1 数据集划分](#4.1 数据集划分)
- [5. 决策树模型构建](#5. 决策树模型构建)
-
- [5.1 确定最优参数组合](#5.1 确定最优参数组合)
- [5.2 模型构建(模型选择、训练、预测)](#5.2 模型构建(模型选择、训练、预测))
- [6. 决策树模型评估](#6. 决策树模型评估)
-
- [6.1 核心评估指标计算](#6.1 核心评估指标计算)
- [6.2 可视化评估](#6.2 可视化评估)
-
- [6.2.1 混淆矩阵可视化](#6.2.1 混淆矩阵可视化)
- [6.2.2 ROC曲线与AUC值可视化](#6.2.2 ROC曲线与AUC值可视化)
- [6.2.3 预测概率分布可视化](#6.2.3 预测概率分布可视化)
- [6.2.4 特征重要性可视化](#6.2.4 特征重要性可视化)
- [6.2.5 决策规则可视化](#6.2.5 决策规则可视化)
一、决策树分类相关理论
决策树(Decision Tree)是一种基于树状结构进行决策的监督学习算法,在分类与回归任务中均有广泛应用,尤其在分类问题中因其逻辑清晰、可解释性强的特点成为经典算法之一。其核心思想是模拟人类决策过程,通过对数据特征的逐步拆分,将复杂的分类问题转化为一系列简单的二元或多元判断,最终形成一棵"根节点-内部节点-叶节点 "的层级结构,其中叶节点直接对应分类结果。
1. 决策树的基本结构
决策树的结构可分为三个核心部分,各节点功能明确且相互关联,共同完成从特征输入到分类输出的全过程:
-
根节点(Root Node):决策树的起点,包含全部待分类的样本数据。根节点的选择直接影响树的复杂度与分类效果,通常通过特征重要性评估确定最优初始拆分特征。
-
内部节点(Internal Node):位于根节点与叶节点之间,每个内部节点对应一个特征判断条件(如"年龄>30""学历=本科"),其作用是根据该特征的取值将父节点的样本集拆分为多个子样本集,实现数据的逐步细化。
-
叶节点(Leaf Node):决策树的终点,每个叶节点代表一个明确的分类结果(如"购买意愿=是""风险等级=低")。当样本数据通过内部节点的层层判断到达叶节点时,即可确定其所属类别。
-
分支(Branch):连接节点的线段,对应特征判断的不同结果,每个分支代表一个子样本集的流向。例如,若内部节点的判断条件为"性别",则分支可分为"男"和"女"两个方向。
核心特点:决策树的结构是"自上而下"生成的,每个节点的拆分过程都是局部最优的选择(基于特定评价准则),最终形成的树结构可直观反映特征与分类结果的关联关系,无需专业背景也能理解决策逻辑。
2. 决策树的分类原理
决策树的分类过程本质是"特征拆分-样本细化-结果确定"的迭代过程,具体可分为两个阶段:树的构建与样本分类。
2.1 树的构建过程(核心阶段)
树的构建遵循"贪心算法"思想,即每次拆分都选择当前最优的特征和判断条件,直至满足停止准则。核心步骤如下:
-
初始化根节点:将全部训练样本纳入根节点,计算所有候选特征的"拆分价值"(通过信息增益、Gini系数等准则评估)。
-
选择最优拆分特征:依据拆分价值准则,筛选出能使子样本集"纯度最高"的特征作为当前节点的判断特征。例如,若通过"是否有房"这一特征拆分后,两个子样本集中"按时还款"和"逾期"的样本分别高度集中,则该特征为最优拆分特征。
-
生成子节点与分支:根据最优特征的不同取值,将当前节点的样本集拆分为多个子样本集,每个取值对应一个分支和一个子节点。
-
迭代拆分:对每个子节点重复步骤1-3,直至满足停止条件(如子样本集全为同一类别、样本数量小于阈值、无可用拆分特征等)。
-
标记叶节点:将停止拆分的子节点标记为叶节点,其类别通常为该节点中样本数量最多的类别(多数投票原则)。
2.2 样本分类过程
对于新的待分类样本,从决策树的根节点开始,根据样本在当前节点特征上的取值,沿着对应的分支向下移动,依次经过内部节点的判断,最终到达叶节点,该叶节点的类别即为样本的分类结果。例如,待分类样本"年龄28岁、本科、有房",从根节点"年龄"开始,沿"25-35岁"分支到下一个节点"学历",再沿"本科"分支到节点"是否有房",最后沿"有房"分支到达叶节点"按时还款=是",完成分类。
3. 关键核心:特征拆分准则
特征拆分准则是决策树构建的"灵魂",其核心目标是通过特征拆分使子样本集的"不确定性降低"(即纯度提升)。常用的拆分准则主要有以下三种:
3.1 信息增益(Information Gain)与ID3算法
信息增益基于"信息论"中的熵(Entropy)概念,熵用于衡量样本集的不确定性,熵值越大,样本集类别越混乱(纯度越低);熵值越小,样本集纯度越高。
- 熵的计算公式 :对于样本集D,假设共有k个类别,第i类别的样本数量为|C|,样本总数为|D|,则熵 H ( D ) H(D) H(D)为:
H ( D ) = − ∑ i = 1 k p i log 2 p i H(D) = -\sum_{i=1}^{k} p_i \log_2 p_i H(D)=−i=1∑kpilog2pi
其中 p = ∣ C ∣ / ∣ D ∣ p = |C| / |D| p=∣C∣/∣D∣,代表第 i i i类样本在 D D D中的占比。当所有样本都属于同一类别时, H ( D ) = 0 H(D)=0 H(D)=0(纯度最高);当样本类别均匀分布时, H ( D ) H(D) H(D)达到最大值(纯度最低)。
- 信息增益的定义 :使用特征A拆分样本集D后,熵的减少量即为信息增益 G ( D , A ) G(D,A) G(D,A),计算公式为:
G ( D , A ) = H ( D ) − ∑ v = 1 V ∣ D v ∣ ∣ D ∣ H ( D v ) G(D,A) = H(D) - \sum_{v=1}^{V} \frac{|D_v|}{|D|} H(D_v) G(D,A)=H(D)−v=1∑V∣D∣∣Dv∣H(Dv)
其中, V V V是特征 A A A的取值个数, D v D_v Dv是 D D D中特征 A A A取值为 v v v的子样本集, ∣ D v ∣ / ∣ D ∣ |D_v|/|D| ∣Dv∣/∣D∣是子样本集的权重。
- ID3算法:以"信息增益最大"为准则选择拆分特征,是最早的决策树算法之一。但该算法存在缺陷------倾向于选择取值较多的特征(如"身份证号"这类唯一值特征,拆分后每个子样本集纯度都为1,信息增益最大,但无实际分类意义)。
3.2 信息增益比(Information Gain Ratio)与C4.5算法
为解决ID3算法的缺陷,C4.5算法引入"信息增益比"作为拆分准则,通过对"取值多的特征"施加惩罚,平衡特征取值数量对拆分选择的影响。
- 信息增益比的计算公式:
G R ( D , A ) = G ( D , A ) H A ( D ) G_R(D,A) = \frac{G(D,A)}{H_A(D)} GR(D,A)=HA(D)G(D,A)
其中 H ( D ) H(D) H(D)是特征 A A A的"固有值"(Intrinsic Value),计算公式为:
H A ( D ) = − ∑ v = 1 V ∣ D v ∣ ∣ D ∣ log 2 ∣ D v ∣ ∣ D ∣ H_A(D) = -\sum_{v=1}^{V} \frac{|D_v|}{|D|} \log_2 \frac{|D_v|}{|D|} HA(D)=−v=1∑V∣D∣∣Dv∣log2∣D∣∣Dv∣
特征 A A A的取值越多, H ( D ) H(D) H(D)越大,信息增益比的惩罚效果越强,从而避免算法优先选择取值过多的无效特征。C4.5算法在ID3基础上还支持连续值特征(通过离散化处理,如将"年龄"划分为多个区间)和缺失值处理(用样本权重弥补缺失数据的影响),实用性更强。
3.3 Gini系数与CART算法
Gini系数(基尼不纯度)衡量的是从样本集中随机抽取两个样本,其类别不同的概率,取值范围为0,1。Gini系数越小,样本集纯度越高(随机抽取的两个样本类别相同的概率越大)。
- Gini系数的计算公式:对于样本集D,Gini系数Gini(D)为:
G i n i ( D ) = 1 − ∑ i = 1 k p i 2 Gini(D) = 1 - \sum_{i=1}^{k} p_i^2 Gini(D)=1−i=1∑kpi2
使用特征 A A A拆分后,子样本集的加权Gini系数为:
G i n i ( D , A ) = ∑ v = 1 V ∣ D v ∣ ∣ D ∣ G i n i ( D v ) Gini(D,A) = \sum_{v=1}^{V} \frac{|D_v|}{|D|} Gini(D_v) Gini(D,A)=v=1∑V∣D∣∣Dv∣Gini(Dv)
CART(Classification and Regression Tree)算法以"Gini系数最小"为准则选择拆分特征,且仅生成二叉树(无论特征取值多少,都拆分为两个子节点,如将"学历"拆分为"本科及以上"和"本科以下"),结构更简洁,计算效率更高,同时支持分类和回归任务,是目前应用最广泛的决策树算法之一。
4. 决策树的过拟合与剪枝策略
决策树在构建过程中若无限制拆分,会导致树结构过于复杂(节点多、深度大),模型会"死记硬背"训练集中的噪声数据和个别异常样本,出现"过拟合"现象------在训练集上分类准确率极高,但在新的测试集上表现极差。解决过拟合的核心方法是"剪枝",通过移除部分冗余节点,简化树结构,提升模型的泛化能力。剪枝策略主要分为两类:
4.1 预剪枝(Pre-pruning)
在树的构建过程中提前终止拆分,从源头避免过拟合。常用的预剪枝条件包括:
-
子样本集的样本数量小于设定阈值(如少于5个样本则停止拆分);
-
当前节点的拆分增益(信息增益、Gini系数提升量)小于设定阈值,即拆分带来的纯度提升不明显;
-
决策树的深度达到设定上限,停止进一步拆分。
预剪枝的优点是计算效率高(无需构建完整树),缺点是可能因"提前停止"导致模型欠拟合(未充分学习数据特征)。
4.2 后剪枝(Post-pruning)
先构建完整的决策树(允许过拟合),再从叶节点向根节点反向修剪,移除对模型泛化能力无贡献的分支。修剪的判断依据通常是"交叉验证误差"------若移除某个子树后,模型在验证集上的分类误差降低,则保留修剪后的结构。
常见的后剪枝方法包括"错误率降低剪枝(REP)""成本复杂度剪枝(CCP)"等。后剪枝的优点是模型泛化能力更强(充分学习后再优化),缺点是计算成本高(需构建完整树并反向验证)。
5. 决策树分类的优缺点
决策树分类的核心优势使其在实际场景中被广泛应用,同时也存在一定局限性,需结合具体任务选择使用。
5.1 优点
-
可解释性极强:树结构直观反映特征与分类结果的逻辑关系,可直接转化为"if-else"规则,便于业务人员理解和验证(如信贷风控中,可清晰解释"为何判定该用户为高风险")。
-
无需特征预处理:对特征的尺度(如"收入"的单位是元还是万元)、类型(连续值、离散值)不敏感,无需进行标准化、归一化处理,减少数据预处理成本。
-
计算效率高:树构建过程为贪心迭代,分类时仅需沿分支遍历,时间复杂度低,适合处理大规模数据。
-
鲁棒性较强:对噪声数据有一定容忍度,少量异常样本不会显著影响整体树结构。
5.2 缺点
-
易过拟合:无剪枝情况下,易因学习训练集噪声导致泛化能力差,需结合剪枝策略优化。
-
对特征变化敏感:少量特征数据的变动可能导致树结构大幅变化,模型稳定性不足。
-
类别不平衡问题:若训练集中某类样本占比极高,决策树可能倾向于优先选择对该类别有利的特征,导致少数类样本分类准确率低。
-
局部最优陷阱:贪心算法的"局部最优选择"无法保证最终树结构为"全局最优",可能存在更优的拆分组合未被选中。
6. 总结
决策树分类是基于树状结构的贪心算法,通过"特征拆分-样本细化"实现分类,核心在于基于信息增益、Gini系数等准则选择最优拆分特征,并通过剪枝策略平衡过拟合与欠拟合。其极强的可解释性和低预处理成本使其在金融、医疗、零售等领域的分类任务中占据重要地位,同时也可作为集成学习算法(如随机森林、XGBoost)的基础组件,进一步提升模型性能。在实际应用中,需结合数据特点选择合适的决策树算法(ID3/C4.5/CART)和剪枝策略,充分发挥其优势并规避局限性。
二、scikit-learn中决策树分类相关方法介绍
scikit-learn(简称sklearn)为决策树分类任务提供了体系化的工具支持,涵盖模型构造、对象操作、结果评估等全流程。本文将围绕决策树分类的构造方法、返回对象的方法与属性,以及核心评估指标展开,帮助开发者系统掌握其在sklearn中的实战应用。
1. 决策树分类构造方法
sklearn中决策树分类的核心构造方法通过sklearn.tree.DecisionTreeClassifier类实现,该方法封装了CART算法(默认),支持通过参数配置实现特征拆分、剪枝等功能,初始化后即可得到决策树分类器对象。
1.1 基础构造语法
python
from sklearn.tree import DecisionTreeClassifier
# 基础构造(使用默认参数)
dt_clf = DecisionTreeClassifier()
# 带参数的构造(常用配置)
dt_clf = DecisionTreeClassifier(
criterion='gini', # 特征拆分准则
max_depth=4, # 树的最大深度(预剪枝)
min_samples_leaf=3, # 叶节点最小样本数(预剪枝)
ccp_alpha=0.01, # 成本复杂度剪枝系数(后剪枝)
class_weight='balanced', # 类别权重(处理不平衡数据)
random_state=42 # 随机种子(保证结果可复现)
)1.2 核心构造参数详解
1.2.1 拆分准则参数
-
criterion :指定特征拆分的评估标准,默认值为
'gini'(Gini系数),可选'entropy'(信息增益)和'log_loss'(对数损失)。数据类别均衡时优先选'gini'(计算高效);类别不平衡或需精准概率输出时,可尝试'entropy'或'log_loss'。 -
splitter :节点拆分策略,默认
'best'(选择当前最优特征),可选'random'(随机选部分特征后再找最优)。小数据集用'best'保证精度;大数据集或集成学习中用'random'引入随机性,减少过拟合。
1.2.2 结构控制参数(预剪枝)
-
max_depth :树的最大深度,默认
None(无限制),是控制过拟合的核心参数。调优建议:从3开始尝试,逐步增加至验证集准确率峰值,常见最优范围3-10。 -
min_samples_split:节点可拆分的最小样本数,默认2。调优建议:增大至5-20,过滤噪声小节点,限制过度拆分。
-
min_samples_leaf:叶节点的最小样本数,默认1。调优建议:设为3-10,避免模型学习个别异常样本,对过拟合控制效果更显著。
-
max_features :拆分时可考虑的最大特征数,默认
None(使用全部特征),可选整数、浮点数、'sqrt'(根号下总特征数)、'log2'。调优建议:高维数据(特征>100)选'sqrt'或'log2',减少冗余提升效率。
1.2.3 正则化与其他参数
-
ccp_alpha :成本复杂度剪枝(CCP)系数,默认0.0(不后剪枝),sklearn中后剪枝的核心参数。调优建议:通过
cost_complexity_pruning_path()获取最优范围,通常取0.01-0.1,值越大剪枝力度越强。 -
class_weight :类别权重,默认
None(等权重),可选'balanced'(按类别频率加权)或自定义字典。适用场景:解决类别不平衡问题(如欺诈检测),提升少数类分类精度。 -
random_state :随机种子,默认
None(结果随机),设为固定整数(如42)可保证模型训练可复现。
注意事项:sklearn的DecisionTreeClassifier默认仅支持CART二叉树,若需ID3/C4.5多叉树,需自定义拆分准则或借助第三方库。
1.3 构造前的环境准备
使用前需安装依赖库,核心命令如下:
bash
# 核心依赖(sklearn+数据处理+可视化)
pip install scikit-learn numpy pandas matplotlib -i https://mirrors.aliyun.com/pypi/simple
# 可选:高质量可视化工具Graphviz
pip install graphviz -i https://mirrors.aliyun.com/pypi/simple2. 决策树分类构造方法返回对象的方法
通过DecisionTreeClassifier构造得到的决策树分类器对象(如上述dt_clf),提供了一系列方法用于模型训练、预测、评估及可视化,覆盖从训练到应用的全流程。
2.1 核心功能方法
| 方法名称 | 语法示例 | 功能说明 |
|---|---|---|
| fit() | dt_clf.fit(X_train, y_train) | 核心训练方法,用训练集(X_train特征矩阵,y_train目标变量)训练模型,无返回值,直接更新分类器对象状态。 |
| predict() | y_pred = dt_clf.predict(X_test) | 预测方法,输入测试集特征矩阵X_test,返回预测的类别标签(数组形式),是模型应用的核心方法。 |
| predict_proba() | proba = dt_clf.predict_proba(X_test) | 概率预测方法,返回每个样本属于各类别的概率值(形状为样本数, 类别数),便于需要概率输出的场景(如风险评分)。 |
| score() | acc = dt_clf.score(X_test, y_test) | 简单评估方法,计算模型在数据集上的准确率(正确分类样本数/总样本数),返回值为0-1的浮点数。 |
| get_depth() | depth = dt_clf.get_depth() | 获取训练后决策树的深度,返回整数,用于模型结构分析。 |
| get_n_leaves() | leaves = dt_clf.get_n_leaves() | 获取训练后决策树的叶节点数量,返回整数,辅助判断模型复杂度。 |
| cost_complexity_pruning_path() | path = dt_clf.cost_complexity_pruning_path(X_train, y_train) | 生成成本复杂度剪枝的路径信息,包含不同alpha值对应的不纯度,用于选择最优ccp_alpha参数。 |
2.2 模型可视化与解释方法
这类方法需结合sklearn.tree模块的辅助函数,实现模型逻辑的可视化呈现,提升可解释性。
| 方法/函数 | 语法示例 | 功能说明 |
|---|---|---|
| export_text()(辅助函数) | from sklearn.tree import export_textrules = export_text(dt_clf, feature_names=feature_list) | 将训练后的决策树转化为文本格式的决策规则,直观展示分类逻辑,便于业务理解。 |
| plot_tree()(辅助函数) | from sklearn.tree import plot_treeplot_tree(dt_clf, filled=True, feature_names=feature_list) | 基于matplotlib绘制决策树结构,支持填充颜色、显示特征/类别名称,生成基础可视化图。 |
| export_graphviz()(辅助函数) | from sklearn.tree import export_graphvizexport_graphviz(dt_clf, out_file='tree.dot', feature_names=feature_list) | 将决策树导出为Graphviz格式文件,结合Graphviz工具可生成高质量、可交互的树状图。 |
3. 决策树分类构造方法返回对象的属性
决策树分类器对象在训练后会生成一系列属性,这些属性存储了模型的核心信息,如特征重要性、树结构参数等,可用于模型分析与解释。所有属性均为"只读",需在模型fit()训练后才能获取有效值。
| 属性名称 | 数据类型 | 功能说明 |
|---|---|---|
| feature_importances_ | 数组(形状为特征数,) | 存储每个特征的重要性得分,得分越高表示该特征对分类结果的贡献越大,是特征筛选的重要依据。 |
| classes_ | 数组(形状为类别数,) | 存储模型识别的类别标签,按升序排列,与predict_proba()返回的概率顺序对应。 |
| n_classes_ | 整数 | 模型识别的类别数量,如鸢尾花数据集分类任务中该值为3。 |
| n_features_ | 整数 | 训练模型时使用的特征数量,与输入X_train的特征数一致。 |
| n_samples_ | 整数 | 训练模型时使用的样本总数,与X_train的样本数一致。 |
| tree_ | Tree对象 | 存储决策树的底层结构信息(如节点类型、拆分特征、阈值等),适合高级用户进行底层分析,一般结合tree_的子属性使用(如tree_.feature存储各节点的拆分特征索引)。 |
4. 决策树分类模型评估指标
模型评估是判断决策树分类效果的核心环节,需结合任务场景选择合适的指标。sklearn的sklearn.metrics模块提供了完整的评估工具,常用指标可分为"基础指标""综合指标"和"类别不平衡指标"三类。
4.1 基础评估指标(混淆矩阵衍生指标)
这类指标基于混淆矩阵(Confusion Matrix)计算,清晰反映模型在各类别上的分类正误情况,适用于二分类和多分类任务。
-
准确率(Accuracy):正确分类的样本数占总样本数的比例,适用于类别均衡的场景。局限性:类别不平衡时失效(如99%样本为负类,随机猜测准确率也达99%)。
-
精确率(Precision):预测为正类的样本中,实际为正类的比例,关注"预测正类的可靠性"(如垃圾邮件识别中,避免正常邮件被误判为垃圾邮件)。
-
召回率(Recall):实际为正类的样本中,被正确预测的比例,关注"正类样本的覆盖度"(如癌症诊断中,避免漏诊真实患者)。
-
F1分数(F1-Score):精确率与召回率的调和平均数,平衡两者的矛盾,适用于需要综合两者的场景。
4.2 综合评估指标
这类指标从整体上评估模型性能,包含多类别适配和概率输出相关的指标。
-
分类报告(Classification Report):一次性输出各类别的精确率、召回率、F1分数及支持样本数,是多分类任务的核心评估工具。
-
对数损失(Log Loss):基于模型的概率输出(predict_proba()结果)计算,衡量预测概率与真实标签的差距,值越小模型概率预测越精准。
4.3 类别不平衡专用指标
当数据中各类别样本占比差异大(如欺诈检测中欺诈样本<5%),需使用以下指标避免传统指标的误导。
-
ROC曲线与AUC值:ROC曲线以假正例率(FPR)为横轴,真正例率(TPR=召回率)为纵轴,AUC值是曲线下面积(0-1),AUC越大模型区分能力越强。适用于二分类任务,多分类需转化为"一对多"或"一对一"模式。
-
马修斯相关系数(MCC):综合考虑混淆矩阵的四个元素,取值范围-1,1,1表示完美分类,0表示随机分类,-1表示完全反置,适合极端不平衡数据。
5. 常见问题与解决方案
在使用sklearn决策树分类时,常遇到过拟合、类别不平衡、可视化失败等问题,以下为针对性解决方案:
5.1 问题1:模型过拟合(训练集准确率高,测试集准确率低)
核心原因:树结构过于复杂,学习了训练集噪声。
解决方案:
-
预剪枝:增大max_depth(如从10减至5)、min_samples_split(如从2增至5)、min_samples_leaf(如从1增至3);
-
后剪枝:增大ccp_alpha值(通过cost_complexity_pruning_path获取最优范围);
-
引入随机性:设置splitter='random'或max_features='sqrt',减少特征冗余;
-
数据层面:增加训练数据量,或通过数据增强减少噪声。
5.2 问题2:类别不平衡(少数类样本分类准确率低)
核心原因:模型倾向于优先学习多数类特征。
解决方案:
-
设置class_weight='balanced',自动给少数类样本加权;
-
数据层面:对少数类过采样(如SMOTE算法)或多数类欠采样;
-
评估指标:重点关注少数类的recall(召回率),而非单纯依赖准确率。
5.3 问题3:可视化失败(plot_tree无输出或export_graphviz报错)
核心原因:matplotlib环境配置问题,或Graphviz工具未正确安装。
解决方案:
-
plot_tree问题:确保matplotlib版本≥3.0,运行前执行plt.show();
-
Graphviz问题:除了pip安装graphviz库,还需安装系统级客户端(如Windows下载安装包后,将安装目录下的bin文件夹添加到系统环境变量);
-
替代方案:使用export_text导出文本规则,无需可视化工具即可查看模型逻辑。
5.4 问题4:模型欠拟合(训练集与测试集准确率均低)
核心原因:树结构过于简单,未充分学习数据特征。
解决方案:
-
减小预剪枝力度:减小max_depth(如从3增至6)、减小min_samples_split(如从5减至2);
-
取消后剪枝:设置ccp_alpha=0.0;
-
特征层面:增加有效特征,或对现有特征进行组合(如将"花瓣长度"与"花瓣宽度"相乘生成新特征)。
三、基于决策树的威斯康星乳腺癌数据分析与挖掘
威斯康星乳腺癌数据集(Wisconsin Breast Cancer Dataset)是医疗数据挖掘领域的经典数据集,包含乳腺肿瘤的细胞核特征指标及病理诊断结果。本文将以该数据集为研究对象,基于scikit-learn的决策树分类器,完成从数据探索、模型构建到性能优化的全流程分析挖掘,最终实现肿瘤良恶性的精准识别,为临床辅助诊断提供参考。
1. 分析目标与技术路线
1.1 核心目标
-
挖掘乳腺肿瘤细胞核特征与良恶性(目标变量)的关联关系;
-
构建基于决策树的分类模型,实现肿瘤良恶性的自动判断;
-
通过参数调优与模型解释,提升模型泛化能力与临床可解释性。
1.2 技术路线
本次分析遵循"数据准备→探索性分析→模型构建(模型选择、参数调优、训练、预测)→模型评估"的核心流程。
2. 数据收集与加载
2.1 威斯康星乳腺癌数据集介绍
2.1.1 数据集基本信息
威斯康星乳腺癌数据集由美国威斯康星大学的研究人员收集,包含乳腺肿块的细针穿刺(FNA)活检数据,主要用于良性/恶性肿瘤的二分类任务。
- 来源:由Dr. William H. Wolberg于1995年公开,现收录于UCI机器学习仓库和Scikit-learn内置数据集;
- 样本量:569个样本(每个样本代表一个乳腺肿块的活检结果);
- 类别分布 :
- 良性(Benign):357个样本(占比约62.7%);
- 恶性(Malignant):212个样本(占比约37.3%);
- 特征类型:全部为连续型数值特征,基于肿块细胞核的图像特征提取;
- 任务目标:根据细胞核特征预测肿瘤为良性(0)或恶性(1)。
2.1.2 数据集字段说明
威斯康星乳腺癌数据集包含31个字段,其中1个为目标标签,30个为特征字段(按"均值""标准误差""最差值"三类分组),具体如下:
| 类别 | 字段英文名称 | 字段中文说明 | 描述 |
|---|---|---|---|
| 目标变量 | diagnosis | 诊断结果 | 0=恶性(Malignant), 1=良性(Benign) |
| 均值特征 | mean radius | 平均半径 | 细胞核半径的算术平均值 |
| mean texture | 平均纹理 | 细胞核纹理的灰度值标准差 | |
| mean perimeter | 平均周长 | 细胞核周长的算术平均值 | |
| mean area | 平均面积 | 细胞核面积的算术平均值 | |
| mean smoothness | 平均平滑度 | 半径长度的局部变化 | |
| mean compactness | 平均紧凑度 | 周长² / 面积 - 1.0 | |
| mean concavity | 平均凹度 | 轮廓凹部分的严重程度 | |
| mean concave points | 平均凹点 | 轮廓凹部分的数量 | |
| mean symmetry | 平均对称性 | 细胞核的对称性 | |
| mean fractal dimension | 平均分形维数 | 海岸线近似 - 1.0 | |
| 标准误差特征 | radius error | 半径误差 | 半径测量的标准误差 |
| texture error | 纹理误差 | 纹理测量的标准误差 | |
| perimeter error | 周长误差 | 周长测量的标准误差 | |
| area error | 面积误差 | 面积测量的标准误差 | |
| smoothness error | 平滑度误差 | 平滑度测量的标准误差 | |
| compactness error | 紧凑度误差 | 紧凑度测量的标准误差 | |
| concavity error | 凹度误差 | 凹度测量的标准误差 | |
| concave points error | 凹点误差 | 凹点测量的标准误差 | |
| symmetry error | 对称性误差 | 对称性测量的标准误差 | |
| fractal dimension error | 分形维数误差 | 分形维数测量的标准误差 | |
| 最差值特征 | worst radius | 最差半径 | 最大的细胞核半径 |
| worst texture | 最差纹理 | 最差的纹理值 | |
| worst perimeter | 最差周长 | 最大的细胞核周长 | |
| worst area | 最差面积 | 最大的细胞核面积 | |
| worst smoothness | 最差平滑度 | 最差的平滑度值 | |
| worst compactness | 最差紧凑度 | 最差的紧凑度值 | |
| worst concavity | 最差凹度 | 最差的凹度值 | |
| worst concave points | 最差凹点 | 最差的凹点值 | |
| worst symmetry | 最差对称性 | 最差的对称性值 | |
| worst fractal dimension | 最差分形维数 | 最差的分形维数值 |
特征统计摘要
| 特征类别 | 特征数量 | 测量内容 |
|---|---|---|
| 均值特征 | 10个 | 细胞核特征的算术平均值 |
| 标准误差特征 | 10个 | 特征测量的变异程度 |
| 最差值特征 | 10个 | 细胞核最异常的特征值 |
这些特征都是从乳腺肿块的细针穿刺(FNA)数字化图像中提取的细胞核特征,用于帮助医生区分恶性肿瘤和良性肿瘤。
2.2 数据加载
Scikit-learn库内置了威斯康星乳腺癌数据集,可直接通过datasets模块加载,避免手动下载和解析文件。以下是加载代码:
python
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from sklearn.datasets import load_breast_cancer
from sklearn.model_selection import train_test_split, GridSearchCV, cross_val_score
from sklearn.tree import DecisionTreeClassifier, plot_tree, export_text
from sklearn.metrics import (confusion_matrix, classification_report,
accuracy_score, precision_score, recall_score, f1_score,
roc_auc_score, roc_curve)
# 设置中文正常显示
plt.rcParams['font.sans-serif'] = ['SimHei']
plt.rcParams['axes.unicode_minus'] = False
# 设置Pandas全局选项
pd.set_option('display.max_columns', None)
pd.set_option('display.width', None)
pd.set_option('display.max_colwidth', 50)
pd.set_option('display.expand_frame_repr', False)
# 加载数据集
cancer = load_breast_cancer()
# 转换为DataFrame便于分析(特征矩阵)
X = pd.DataFrame(data=cancer.data, columns=cancer.feature_names)
# 目标变量(诊断结果)
y = pd.Series(data=cancer.target, name='diagnosis')3. 探索性数据分析(EDA)
探索性数据分析旨在挖掘数据内在规律,为后续模型构建提供依据。重点分析特征分布、特征相关性及良恶性样本的特征差异。
3.1 数据基本信息
3.1.1 查看数据前10行
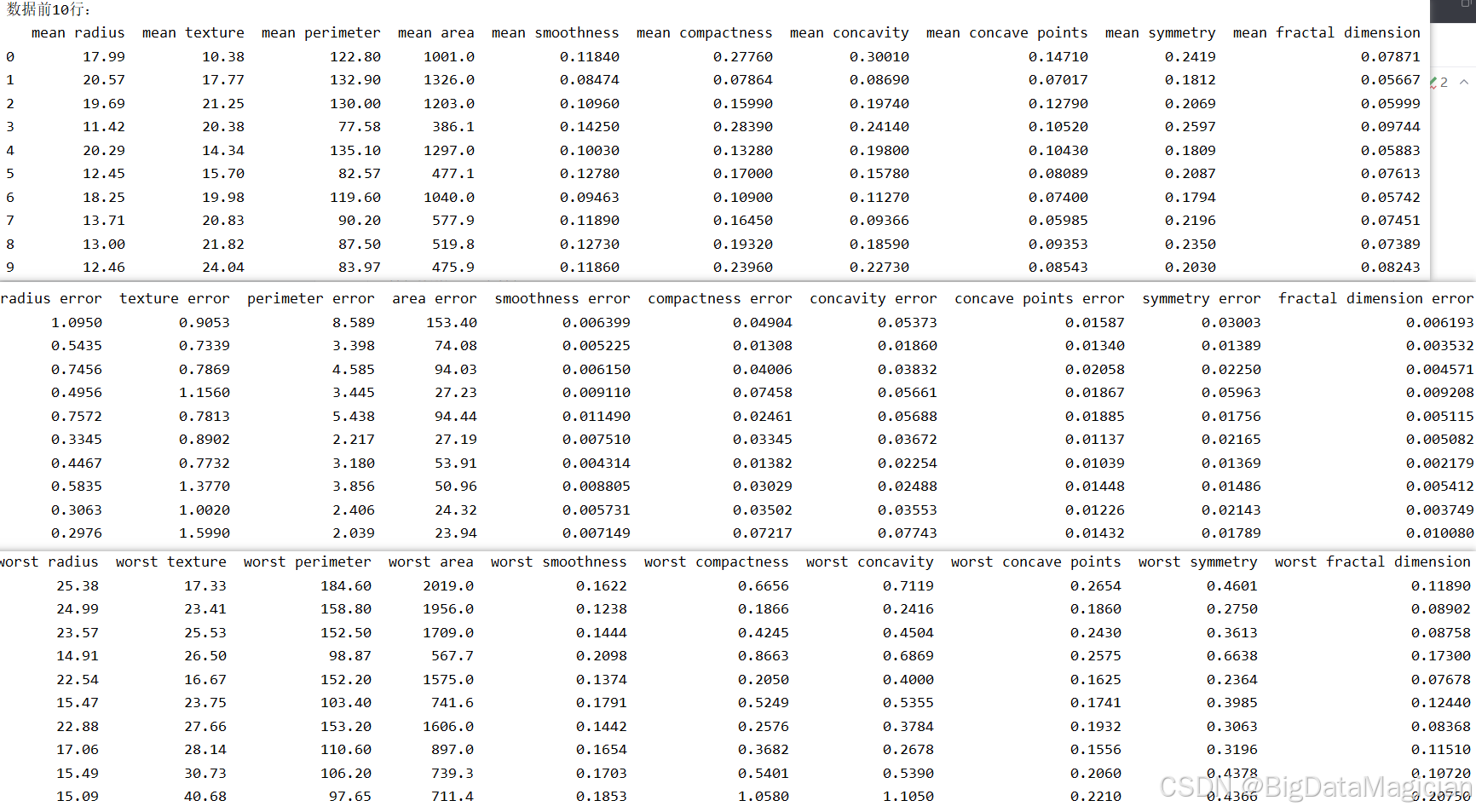
在数据分析的初始阶段,查看数据集的前几行是了解其结构、字段含义和数据类型的重要步骤。通过观察原始数据的分布与格式,可以初步判断是否存在异常值、缺失值或不一致的命名规范,并为后续的数据清洗、特征工程和建模工作提供直观依据。
该代码将特征矩阵 X(包含所有数值型特征)与标签向量 y(类别标签)沿列方向进行拼接,形成一个完整的 DataFrame,便于统一查看样本的完整信息;随后调用 head(10) 输出前10个样本,快速呈现数据结构和内容。
python
# 合并特征和标签,便于同时查看
data = pd.concat([X, y], axis=1) # 按列合并
print("数据前10行:")
print(data.head(10))数据集中各字段部分数据如下图所示,从输出结果可见,数据集中包含多个描述肿瘤形态学特征的数值变量,如 mean radius(平均半径)、mean texture(平均纹理)、mean perimeter(平均周长)等,均为连续型数值。此外还包含误差项(如 radius error)和最差情况指标(如 worst radius),反映肿瘤的异质性与恶化程度。
3.1.2 查看数据摘要信息
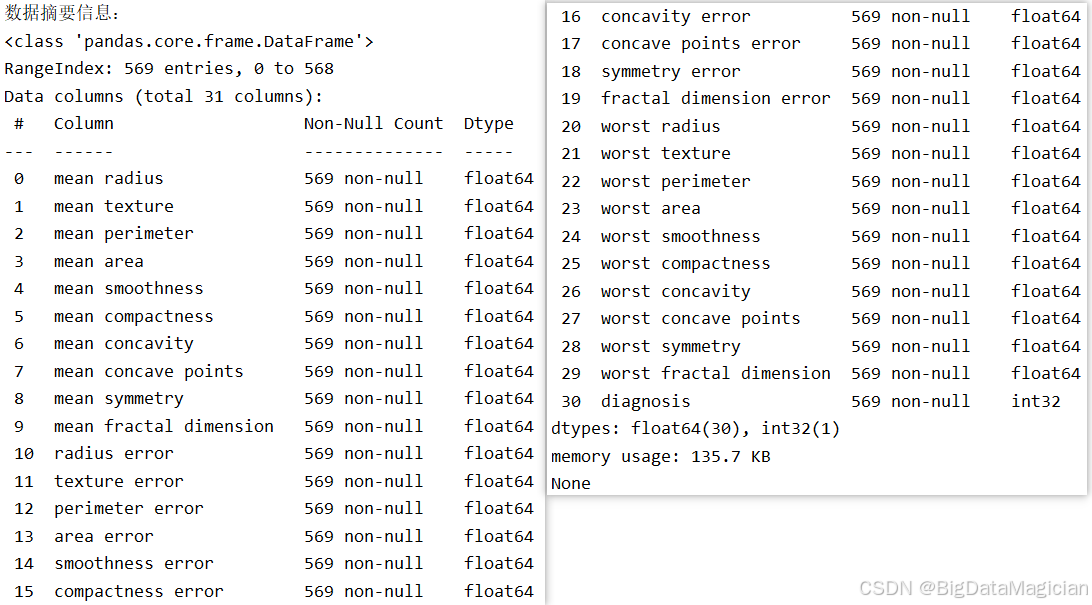
在数据探索阶段,了解数据集的整体结构和基本属性是确保后续分析顺利进行的关键步骤。info() 方法提供了关于 DataFrame 的综合信息,包括样本数量、特征数量、各列的数据类型以及非空值计数,有助于快速判断是否存在缺失值、数据类型是否一致等问题。
该代码调用 Pandas 的 info() 方法,输出数据框的元信息。它会显示数据集的总行数(样本数)、列数(字段数)、每列的名称、非空值数量(Non-Null Count)以及对应的数据类型(Dtype),从而全面反映数据的结构特征。
python
print("\n数据摘要信息:")
print(data.info())从输出结果可见,该数据集共包含 569 个样本 和 31 个字段 ,其中前 30 列为肿瘤形态学特征,均为 float64 类型,表示连续数值;最后一列为标签列 diagnosis,类型为 int32,代表二分类结果(0 表示恶性,1 表示良性)。所有列的 Non-Null Count 均为 569,表明 无任何缺失值,数据完整性高,无需进行缺失值填充或删除操作。此外,数据类型统一且符合预期,无需转换,极大简化了预处理流程,为后续建模提供了高质量的数据基础。
3.1.3 特征描述性统计
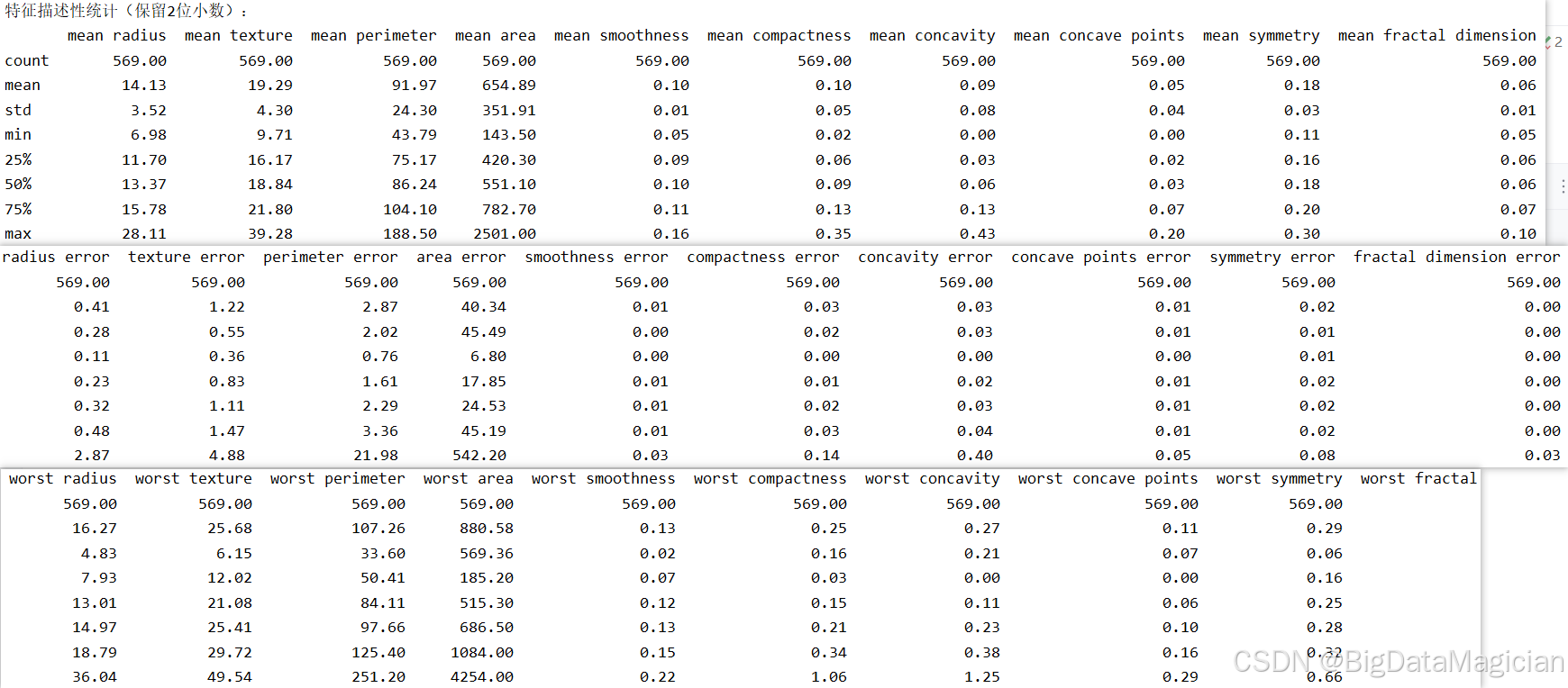
在数据探索过程中,描述性统计分析是理解特征分布特性的核心手段。通过 describe() 方法可以快速获取每个数值型特征的关键统计指标,如均值、标准差、四分位数和极值等,从而评估其集中趋势、离散程度与分布形态。本节对特征矩阵进行描述性统计分析,旨在揭示各特征的数值范围、变异程度及潜在偏态情况,为后续的数据预处理(如标准化)和建模策略提供依据。
该代码调用 Pandas 的 describe() 方法,计算特征矩阵 X 的基本统计量,包括样本数量(count)、均值(mean)、标准差(std)、最小值(min)、25%分位数(25%)、中位数(50%)、75%分位数(75%)和最大值(max)。使用 .round(2) 将结果保留两位小数,提升可读性,便于直观比较不同特征的分布特性。
python
print("\n特征描述性统计(保留2位小数):")
print(X.describe().round(2)) # 仅对特征进行统计,保留2位小数从输出结果可见,30个特征在数值范围和离散程度上存在显著差异。例如,area_worst 最大值达 4254.00,最小值为 135.80,极差超过 4100,而 fractal_dimension_mean 的极差仅为 0.16,表明不同特征的尺度差异极大。这种不一致性会导致距离类算法(如 KNN)在计算样本间相似度时,高尺度特征(如面积)主导距离贡献,而低尺度特征(如纹理误差)被忽略,造成模型偏差。此外,部分特征的标准差较大(如 area_mean 标准差为 351.91),说明数据波动剧烈;而另一些特征(如 smoothness_se 标准差仅 0.01)则高度集中。分布形态方面,多数特征的均值与中位数接近(如 radius_mean 均值 14.13 vs 中位数 13.37),呈近似对称分布;但也有特征如 area_se 均值(56.14)远大于中位数(24.53),呈现明显右偏,提示存在异常值或长尾分布。综上,这些特征需进行标准化处理以消除尺度影响,并结合偏态情况考虑是否进行对数变换等进一步预处理。
3.2 目标变量类别分布分析
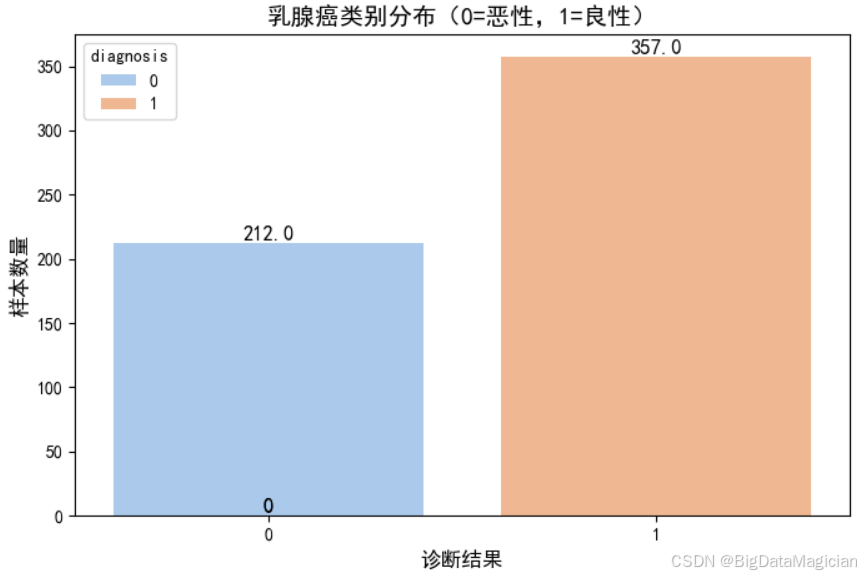
类别分布分析是分类任务中至关重要的探索性步骤,用于评估目标变量的样本比例是否均衡。若某一类别样本显著少于其他类别,则可能导致模型偏向多数类,影响少数类的识别能力。本节通过可视化手段展示乳腺癌数据集中良性与恶性肿瘤的样本数量分布,判断是否存在类别不平衡问题,并为后续建模策略(如是否采用过采样、欠采样或调整类别权重)提供依据。
该代码使用 Seaborn 的 countplot 绘制目标变量 y 的频数分布图,其中 x=y 表示以诊断结果为分类变量。通过设置中文字体和样式增强可读性,同时在每个柱子上方添加具体数值标签,直观显示各类别样本数量。最终将图表保存为图像文件,便于报告展示。
python
import matplotlib.pyplot as plt
import seaborn as sns
# 设置可视化风格
plt.rcParams['font.sans-serif'] = ['SimHei']
plt.rcParams['axes.unicode_minus'] = False
print("\n目标变量类别分布:")
print(y.value_counts())
# 绘制类别分布柱状图
plt.figure(figsize=(8, 5))
ax = sns.countplot(x=y, hue=y, palette='pastel')
ax.set_title('乳腺癌类别分布(0=恶性,1=良性)', fontsize=14)
ax.set_xlabel('诊断结果', fontsize=12)
ax.set_ylabel('样本数量', fontsize=12)
# 添加数值标签
for p in ax.patches:
ax.text(s=p.get_height(), x=p.get_x() + p.get_width() / 2., y=p.get_height(), ha='center', va='bottom', fontsize=12)
plt.savefig('目标变量类别分布.png')从生成的柱状图可见,良性肿瘤(标签为 1)样本数量为 357 个,恶性肿瘤(标签为 0)样本数量为 212 个,两类样本比例约为 1.7:1。虽然良性样本略多,但未出现极端不平衡(如 9:1 或更高),属于机器学习中可接受的范围。在此比例下,大多数分类算法(如逻辑回归、随机森林)仍能有效学习两类特征差异,无需额外处理类别不平衡问题。
3.3 目标变量类别与特征相关性分析
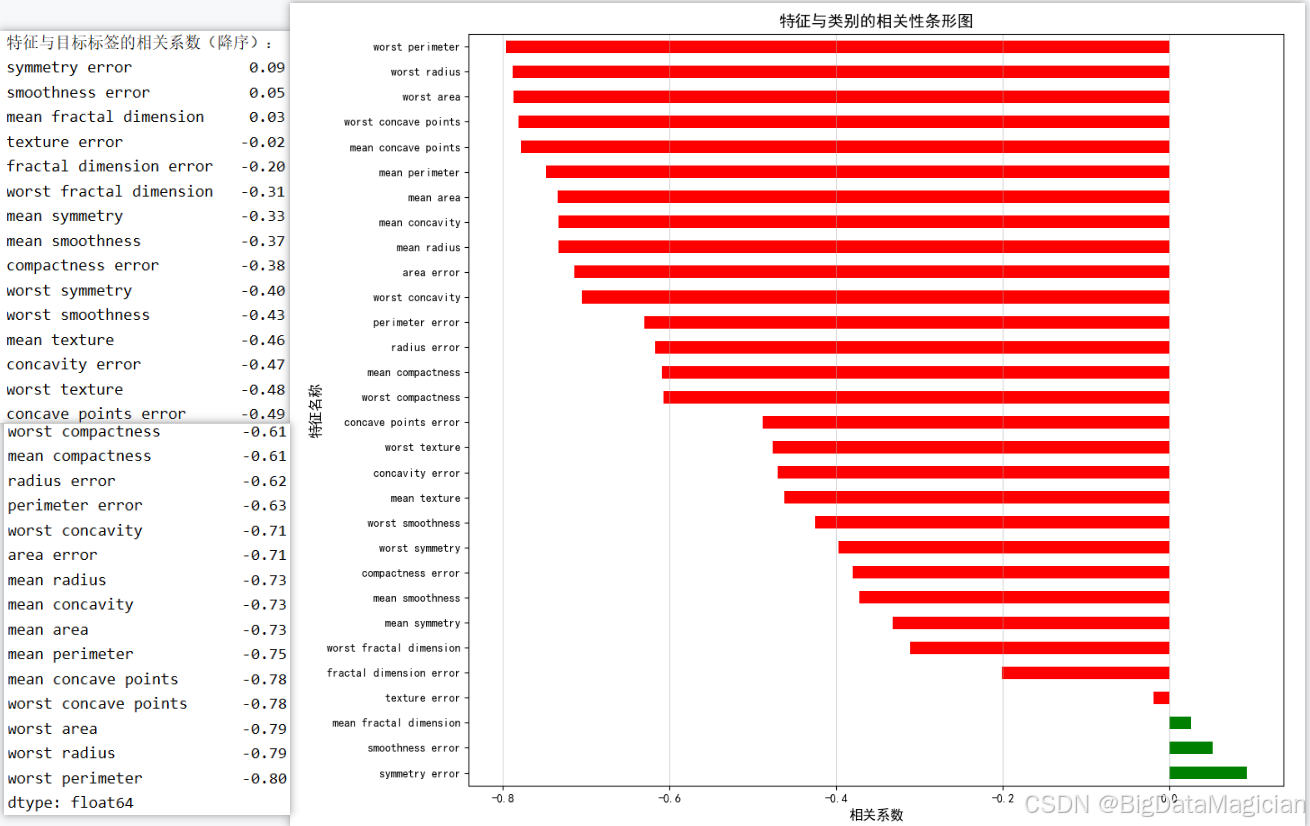
在分类任务中,理解各特征与目标变量之间的关联强度有助于识别对预测结果影响最大的关键特征。本节通过计算每个特征与诊断标签(良性/恶性)之间的斯皮尔曼相关系数,评估其单调关系的强弱,从而筛选出具有高判别能力的特征。由于目标变量为二分类整数标签(0/1),使用非参数的斯皮尔曼相关系数更为稳健,能有效捕捉非线性趋势。结合可视化排序,可直观呈现特征的重要性,为后续特征选择和模型优化提供依据。
该代码首先调用 X.corrwith(y, method='spearman') 计算每个特征与标签之间的斯皮尔曼相关系数,并按降序排列;随后绘制水平条形图,正相关系数以绿色表示,负相关以红色表示,便于快速识别关键特征。图形清晰展示各特征对分类任务的贡献方向与强度。
python
# 计算每个特征与目标标签的相关系数(斯皮尔曼相关系数)
feature_label_corr = X.corrwith(y, method='spearman').sort_values(ascending=False)
print("特征与目标标签的相关系数(降序):")
print(feature_label_corr.round(2))
# 绘制水平柱状图
plt.figure(figsize=(12, 10))
colors = ['green' if x > 0 else 'red' for x in feature_label_corr.values]
feature_label_corr.plot(kind='barh', color=colors)
plt.title('特征与类别的相关性条形图', fontsize=14)
plt.xlabel('相关系数', fontsize=12)
plt.ylabel('特征名称', fontsize=12)
plt.tight_layout()
plt.grid(axis='x', alpha=0.5)
plt.savefig('特征与类别相关性排序.png')从输出结果可见,多数特征与诊断标签呈显著负相关,其中 worst perimeter、worst radius、worst area 等"最差"系列特征相关系数接近 -0.8,表明其数值越大,越倾向于恶性肿瘤,是区分良恶性的最强指标。而 mean concavity、mean concave points、worst concavity 等反映边缘不规则性的特征也表现出较强负相关(约 -0.7~ -0.8),进一步验证了恶性肿瘤形态更不规则的医学规律。少数特征如 symmetry error、smoothness error 相关系数接近 0,表明其对分类贡献较小。整体来看,尺寸和凹陷类特征主导分类过程,在KNN建模时应优先保留这些高相关特征,剔除低相关项以提升模型效率与准确性。
3.4 关键特征分布分析
在分类任务中,理解关键特征在不同类别之间的分布差异对于模型构建具有重要意义。本节通过绘制核密度估计图(Kernel Density Estimate, KDE) ,直观展示多个与诊断结果高度相关的特征在良性(diagnosis=1)和恶性(diagnosis=0)两类样本中的概率密度分布情况。KDE是一种非参数方法,能够平滑地估计数据的概率密度函数,从而揭示数据的分布形态、集中趋势及分离程度。
从医学角度出发,乳腺肿瘤的良恶性往往与其细胞核的形态学异常程度密切相关:恶性肿瘤通常表现出更大的尺寸、更高的不规则性(如凹陷、边缘粗糙等)。因此,我们重点关注那些与"最差"(worst)或"凹度"(concavity)相关的特征,这些特征在先前的相关性分析中已显示出较强的判别能力。通过观察这些特征在两类样本中的分布是否明显分离,可以判断其作为分类依据的有效性,并为后续特征选择和模型训练提供支持。
python
# 选择关键特征
key_features = feature_label_corr[abs(feature_label_corr) > 0.5].index.tolist()
# 绘制核密度图,KDE:核密度估计
plt.figure(figsize=(18, 12))
feature = key_features
for i in range(len(feature)):
plt.subplot(3, 5, i + 1)
sns.kdeplot(
data=data,
x=feature[i],
hue='diagnosis',
fill=True,
common_norm=False, # 每个类别单独归一化
alpha=0.5
)
plt.title(f'{feature[i]} 分布')
plt.xlabel(feature[i])
plt.tight_layout()
plt.savefig('乳腺癌关键特征KDE分布.png')从生成的核密度图可以看出,多个关键特征在良性与恶性肿瘤之间呈现出明显的分布差异:
-
worst_perimeter、worst_radius、worst_area:这三个"最差"系列的尺寸特征中,恶性肿瘤(蓝色)的密度峰值明显偏向更高数值区间,而良性肿瘤(橙色)集中在较低值区域。这表明恶性肿瘤的细胞核通常更大、更异常,具备更强的区分度。 -
mean_concavity、worst_concavity、mean_concave_points、worst_concave_points:反映细胞核边缘凹陷程度的特征,在恶性样本中表现出更高的值和更宽的分布范围,且与良性样本的分布几乎无重叠,说明这些特征对识别恶性肿瘤极为敏感。 -
mean_compactness、worst_compactness:紧凑度越高表示形状越不规则,恶性肿瘤在这两项上的分布也显著偏移至高值区,进一步验证了恶性肿瘤形态异质性强的特点。 -
radius_error、perimeter_error、area_error:误差项反映了测量的波动性,恶性肿瘤在这些指标上也表现出更大的变异性和更高的均值,可能暗示其结构不稳定。
总体来看,大多数高相关特征在两类样本间具有良好的可分性,尤其是在"最差"系列和"凹度"类特征上,分布几乎不重叠,说明它们能有效帮助KNN算法进行邻近点搜索和分类决策。这些发现不仅增强了我们对数据的理解,也为后续建模提供了明确的方向,应优先保留这些具有强判别能力的特征,以提升KNN模型的预测性能。

3.5 关键特征相关性分析
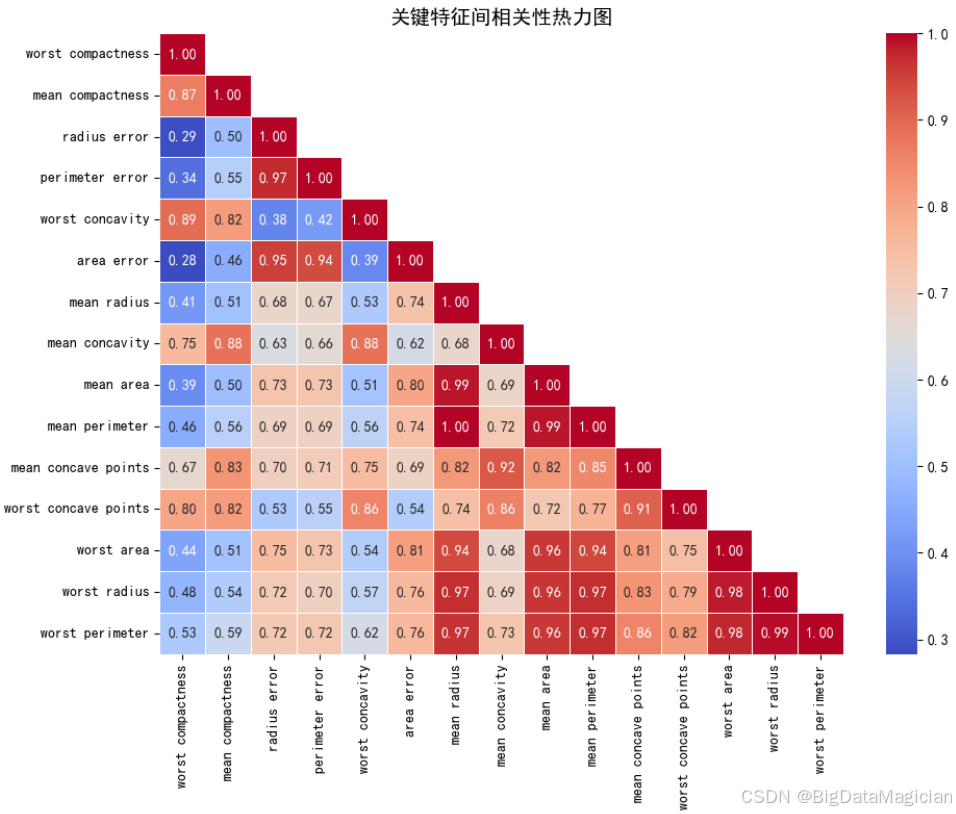
在构建分类模型之前,理解特征之间的相互关系至关重要。高度相关的特征可能导致多重共线性 问题,影响模型的稳定性和解释性,尤其是在基于距离的算法(如KNN)中,冗余特征可能放大某些维度的影响,降低模型泛化能力。因此,本节通过计算关键特征之间的皮尔逊相关系数,并绘制热力图,直观展示各特征间的线性相关程度。
选取了在前序分析中特征与类别相关性绝对值大于0.5的关键特征,这些特征主要反映肿瘤细胞核的尺寸、形状不规则性和边缘凹陷等重要形态学指标。通过分析它们之间的相关性,可以识别出是否存在信息重叠的特征对,从而为后续的特征选择或降维提供依据。例如,若两个特征高度正相关(如 >0.8),则可考虑保留其中一个以减少冗余,提升模型效率与鲁棒性。
python
import numpy as np
# 计算特征间的相关系数(皮尔逊相关系数)
corr_matrix = X[key_features].corr(method='pearson')
# 创建掩码,只保留下三角部分(包括对角线)
mask = np.triu(m=np.ones_like(corr_matrix, dtype=bool), k=1)
# 绘制热力图
plt.figure(figsize=(10, 8))
sns.heatmap(data=corr_matrix, mask=mask, annot=True, cmap='coolwarm', fmt='.2f', linewidths=0.5)
plt.title('关键特征间相关性热力图', fontsize=14)
plt.tight_layout()
plt.savefig('关键特征间相关性热力图.png')从生成的热力图可以看出,所选关键特征之间普遍存在较强的正相关关系,符合生物学逻辑:
-
尺寸类特征高度相关:
worst_radius、worst_perimeter、worst_area三者之间的相关系数均超过 0.97,表明它们在描述肿瘤最大形态时几乎完全一致,属于高度冗余的特征。- 同样,
mean_radius、mean_perimeter、mean_area也表现出较强正相关(>0.7),说明平均尺寸特征也具有一定的信息重叠。
-
凹陷与不规则性特征群:
mean_concavity与mean_concave_points相关系数达 0.82 ,worst_concavity与worst_concave_points达 0.91,说明凹陷程度与凹点数量密切相关,二者共同反映了细胞核边缘的异常程度。- 此外,
mean_compactness与mean_concavity的相关系数为 0.88,进一步验证了"紧凑度"与"凹度"在几何上的对立关系(越凹则越不紧凑)。
-
误差项与主特征相关性较弱:
- 如
radius_error、area_error等虽与其他特征有一定相关性(约0.3~0.5),但整体偏低,说明测量波动性相对独立于总体形态,可作为补充信息保留。
- 如
综上所述,该热力图揭示了多个特征组合之间的强相关性 ,提示我们在后续建模过程中应谨慎处理这些冗余变量。例如,在KNN中,如果同时包含worst_radius和worst_area,由于它们几乎线性相关,可能会导致某一个特征主导距离计算,从而降低模型鲁棒性。
3.6 关键特征的类别区分能力验证
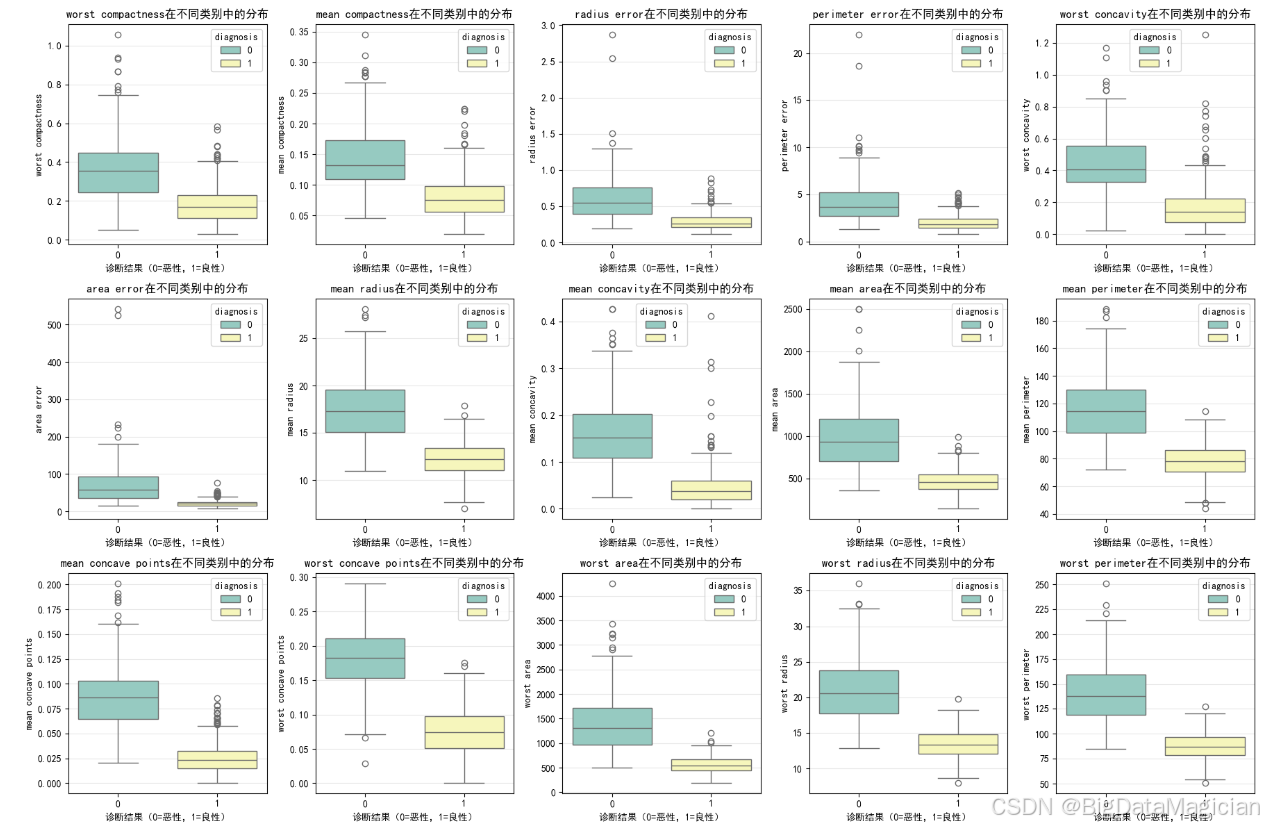
在分类任务中,特征的判别能力(即其在不同类别之间是否存在显著差异)是决定模型性能的关键因素。为了进一步验证前文筛选出的高相关性特征是否具备良好的类别区分效果,本节采用箱线图(Boxplot) 对这些关键特征在良性(diagnosis=1)与恶性(diagnosis=0)两类样本中的分布进行可视化对比。
箱线图能够清晰地展示数据的中位数、四分位距、异常值等统计信息,特别适合用于比较两个或多个组之间的分布差异。通过观察各类别下特征的中心位置(中位数)、离散程度(箱体高度)以及异常值分布,可以直观判断该特征是否能有效将两类样本区分开来。例如,若某一特征在恶性肿瘤中的中位数远高于良性肿瘤,且两组箱体无重叠,则说明该特征具有很强的判别力。
选取了与目标变量斯皮尔曼相关系数绝对值大于0.5的特征作为"关键特征",这些特征已在相关性分析中被确认为对分类结果有较强影响。接下来通过箱线图验证其实际分布上的可分性,从而为后续建模提供更可靠的依据。
python
# 选择关键特征
key_features = feature_label_corr[abs(feature_label_corr) > 0.5].index.tolist()
# 绘制箱线图
plt.figure(figsize=(18, 12))
# enumerate() 函数同时返回索引和元素值,参数 1 表示索引从 1 开始(默认从 0 开始)
for i, feature in enumerate(key_features, 1):
plt.subplot(3, 5, i)
sns.boxplot(x=y, y=data[feature], hue=y, palette='Set3')
plt.title(f'{feature}在不同类别中的分布', fontsize=12)
plt.xlabel('诊断结果(0=恶性,1=良性)', fontsize=10)
plt.ylabel(feature, fontsize=10)
plt.grid(axis='y', alpha=0.3)
plt.tight_layout()
plt.savefig('关键特征类别区分箱线图.png')从箱线图可见,所选关键特征在良性(1)与恶性(0)肿瘤之间均表现出显著的分布差异。例,尤其是在"最差"系列和"凹度"类特征上,箱体几乎没有交集,说明它们对区分良恶性具有极高的判别价值。此外,多数特征的四分位距(IQR)较小,说明数据集中趋势明确,异常值较少。虽然部分特征如 mean radius 存在少量重叠,但总体仍能有效分离两类。这验证了相关性分析的结果,确认这些特征是构建KNN模型的理想输入,可显著提升分类性能。
从图中可见部分特征存在异常值,但这些值源于真实有效的医学观测,反映了肿瘤在形态学上的极端但合理的生物学变异,具有重要的临床意义,因此无需剔除或修正。保留这些样本有助于模型更好地识别高风险恶性病例,提升实际应用中的泛化能力与鲁棒性。
4. 数据预处理
决策树模型对特征尺度不敏感,无需进行标准化或归一化处理,但需确认数据无缺失值和异常值。结合前文X.info()结果,数据集无缺失值,异常值可通过模型参数(如min_samples_leaf)间接控制,因此无需额外预处理。此外,决策树对多重共线性也不敏感,因此也无需进行相关处理。
4.1 数据集划分
为客观验证决策树模型的泛化能力,需将筛选后的特征集与标签集划分为训练集和测试集。考虑到威斯康星乳腺癌数据集存在类别分布特性,若采用随机抽样易导致训练集与测试集类别分布失衡,因此选择分层抽样策略,确保划分后训练集与测试集的类别分布与原始数据集保持一致,避免因样本分布偏差影响模型评估结果的可靠性。
本次划分将数据集按 8:2 的比例分配为训练集与测试集,其中训练集用于模型的参数学习与结构构建,测试集用于独立验证模型的预测性能。同时为保证实验结果的可复现性,设置固定的随机种子以锁定抽样结果。
python
from sklearn.model_selection import train_test_split
X_selected = X[key_features]
# 划分训练集(80%)和测试集(20%)
X_train, X_test, y_train, y_test = train_test_split(
X_selected, y,
test_size=0.2,
random_state=42,
stratify=y # 分层抽样,保持类别分布一致
)
# 查看划分后的数据形状
print(f"训练集特征矩阵形状:{X_train.shape}")
print(f"测试集特征矩阵形状:{X_test.shape}")
print(f"训练集类别分布:\n{y_train.value_counts(normalize=True).round(2)}")
print(f"测试集类别分布:\n{y_test.value_counts(normalize=True).round(2)}")划分完成后对数据规模与类别分布进行验证,如下图所示,训练集样本量约占总样本的 80%,测试集占比 20%,符合预设的划分比例;且训练集与测试集中良性、恶性肿瘤样本的占比完全匹配原始数据集分布,证明分层抽样有效维持了数据的原始类别结构,为后续决策树模型的训练与评估奠定了可靠的数据基础。

5. 决策树模型构建
5.1 确定最优参数组合
为提升决策树模型的泛化能力、避免过拟合,需结合预剪枝与后剪枝策略进行参数调优。首先通过成本复杂度剪枝(CCP)方法获取训练集上的剪枝系数候选值,并筛选出合理范围的系数(排除仅保留根节点的极端情况);同时针对决策树的核心结构参数(拆分准则、最大深度、叶节点最小样本数)构建参数网格,覆盖不同准则、深度与叶节点规模的组合。
考虑到乳腺癌数据集存在类别不平衡特性,选择F1分数作为评估指标(平衡精确率与召回率),并采用5折交叉验证对参数组合进行遍历验证,以确保评估结果的稳定性。同时为进一步缓解类别不平衡影响,在模型训练中设置类别权重平衡策略。
python
# 在训练集上获取所有可能的剪枝点
clf = DecisionTreeClassifier(random_state=42)
path = clf.cost_complexity_pruning_path(X_train, y_train)
# 排除最后一个(只剩根节点)
ccp_alphas = path.ccp_alphas[:-1]
ccp_alphas_filtered = ccp_alphas[(ccp_alphas > 0.01) & (ccp_alphas < 0.1)]
# 定义参数网格
param_grid = {
'criterion': ['gini', 'entropy', 'log_loss'], # 拆分准则
'max_depth': [3, 4, 5, 6, 7], # 树的最大深度(预剪枝)
'min_samples_leaf': [2, 3, 4, 5, 6, 7], # 叶节点最小样本数(预剪枝)
'ccp_alpha': ccp_alphas_filtered # 成本复杂度剪枝系数(后剪枝)
}
# 选择F1分数作为评估指标通常是因为在处理不平衡分类问题时,准确率(Accuracy)可能不是最佳的性能度量标准。准确率仅考虑了正确预测的比例,但在类别分布极不均匀的情况下,仅仅依赖于准确率可能会导致对模型性能的误解。
# 初始化网格搜索(结合5折交叉验证,以F1分数为优化目标)
grid_search = GridSearchCV(
estimator=DecisionTreeClassifier(random_state=42, class_weight='balanced'), # 平衡类别权重,通过给少数类更高权重,让模型在训练时"更在乎"少数类,从而缓解类别不平衡带来的偏差。
param_grid=param_grid,
cv=5,
scoring='f1', # 以F1分数为评估指标(平衡精确率与召回率)
verbose=1
)
# 执行网格搜索
grid_search.fit(X_train, y_train)
# 输出最优参数与最优交叉验证分数
print("最优参数组合:", grid_search.best_params_)
print("最优交叉验证F1分数:", grid_search.best_score_.round(4))参数搜索过程共对270组候选参数组合进行了5折交叉验证,总计完成1350次模型训练。最终得到的最优参数组合兼顾了树的复杂度与拟合能力,对应的5折交叉验证F1分数达到0.9503,证明该参数组合在训练集上具备良好的分类性能,同时通过剪枝与结构约束有效控制了过拟合风险。

5.2 模型构建(模型选择、训练、预测)
基于参数调优得到的最优参数组合,构建最终的决策树分类模型,并在独立测试集上进行预测验证。首先调用网格搜索得到的最优估计器,以其为基础生成最终模型;随后利用该模型对测试集的特征数据进行两类预测,一是直接输出类别标签,二是输出样本属于各类别的概率值,从而更直观地体现模型对预测结果的置信度。
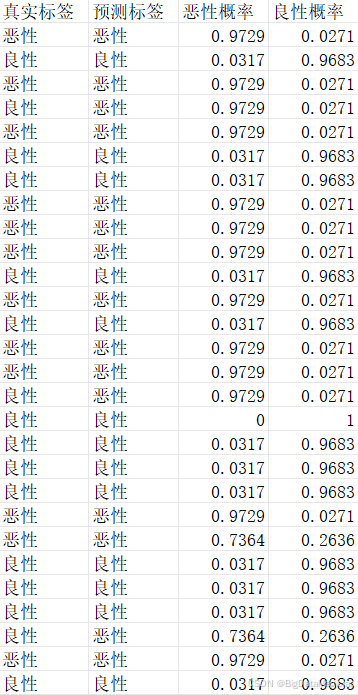
为增强结果的可读性,将测试集的真实标签与模型预测标签均转换为"恶性""良性"的类别名称,再结合对应的类别概率值,整合为包含"真实标签""预测标签""恶性概率""良性概率"的结果表格,并导出为文件留存。
python
# 基于最优参数构建最终模型
dt_best = grid_search.best_estimator_
# 在测试集上评估最终模型
y_pred_best = dt_best.predict(X_test)
y_pred_proba_best = dt_best.predict_proba(X_test)
# 将类别标签转换为类别名称
y_test_names = y_test.reset_index(drop=True).replace({0: '恶性', 1: '良性'})
y_pred_names = pd.Series(y_pred_best).replace({0: '恶性', 1: '良性'})
# 构建测试集预测结果DataFrame
test_pred_df = pd.DataFrame({
'真实标签': y_test_names,
'预测标签': y_pred_names,
'恶性概率': y_pred_proba_best[:, 0].round(4),
'良性概率': y_pred_proba_best[:, 1].round(4)
})
test_pred_df.to_csv('最优模型测试集预测结果及概率.csv', index=False)从部分预测结果可以看到模型对多数样本的预测置信度较高(如恶性样本的恶性概率达0.9729、良性样本的良性概率达0.9683),仅少数样本出现预测偏差,整体呈现出较好的分类区分度,为后续模型性能评估提供了具体的预测依据。
6. 决策树模型评估
6.1 核心评估指标计算
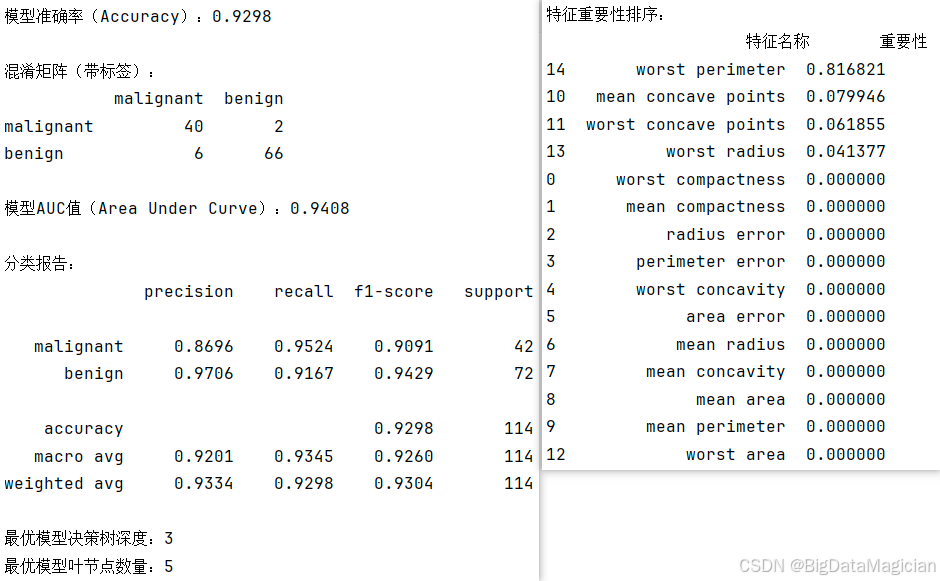
为全面评估最优决策树模型的分类性能,从分类正确性、类别区分度、模型结构及特征贡献度等维度,选取准确率、混淆矩阵、AUC值、分类报告、树结构参数、特征重要性等指标进行量化分析。
首先通过准确率直观反映模型在测试集上的整体预测正确率;借助混淆矩阵明确不同类别(恶性、良性)的真实与预测结果对应关系,清晰呈现模型的误判分布;AUC值则用于衡量模型对两类样本的区分能力,值越接近1代表区分度越强;分类报告进一步细化至每个类别,输出精确率(预测为某类的样本中真实为该类的比例)、召回率(真实为某类的样本中被正确预测的比例)、F1分数(精确率与召回率的调和平均),以适配乳腺癌数据的类别不平衡特性。同时,统计最优决策树的深度与叶节点数量,辅助判断模型的复杂度与过拟合风险;通过特征重要性排序,明确各输入特征对模型分类决策的贡献程度。
python
from sklearn.metrics import (accuracy_score, classification_report, confusion_matrix, roc_auc_score)
import pandas as pd
# 准确率
test_accuracy = accuracy_score(y_test, y_pred_best)
print(f"\n模型准确率(Accuracy):{test_accuracy:.4f}")
# 混淆矩阵(带标签)
cm = confusion_matrix(y_test, y_pred_best)
cm_df = pd.DataFrame(cm, index=cancer.target_names, columns=cancer.target_names)
print("\n混淆矩阵(带标签):")
print(cm_df)
# AUC计算(需预测概率)
auc = roc_auc_score(y_test, y_pred_proba_best[:, 1])
print(f"\n模型AUC值(Area Under Curve):{auc:.4f}")
# 输出详细分类报告(按类别展示指标)
cr = classification_report(y_test, y_pred_best, target_names=cancer.target_names, digits=4)
print("\n分类报告:")
print(cr)
# 最优模型树结构信息
print(f"最优模型决策树深度:{dt_best.get_depth()}")
print(f"最优模型叶节点数量:{dt_best.get_n_leaves()}")
# 特征重要性计算与排序
feature_importance = pd.DataFrame({
'特征名称': key_features,
'重要性': dt_best.feature_importances_
}).sort_values(by='重要性', ascending=False)
print("\n特征重要性排序:")
print(feature_importance)从评估结果可见:
- 模型整体准确率达0.9298,说明近93%的测试样本被正确分类;
- 混淆矩阵显示,42个恶性样本中仅2个被误判为良性,72个良性样本中6个被误判为恶性,误判规模较小;
- AUC值接近0.94,表明模型对两类样本的区分能力较强;
- 分类报告中,恶性类别的F1分数达0.9091,良性类别的F1分数达0.9429,两类性能均衡;
- 决策树深度为3、叶节点数量为5,属于较简洁的树结构,降低了过拟合风险;
- 特征重要性排序显示,"worst perimeter"(最差周长)是影响分类决策的核心特征,其重要性远超其他特征,其余多数特征对模型决策无贡献。
这些指标共同表明,该决策树模型在威斯康星乳腺癌数据集上具备良好的分类性能与泛化能力。
6.2 可视化评估
6.2.1 混淆矩阵可视化
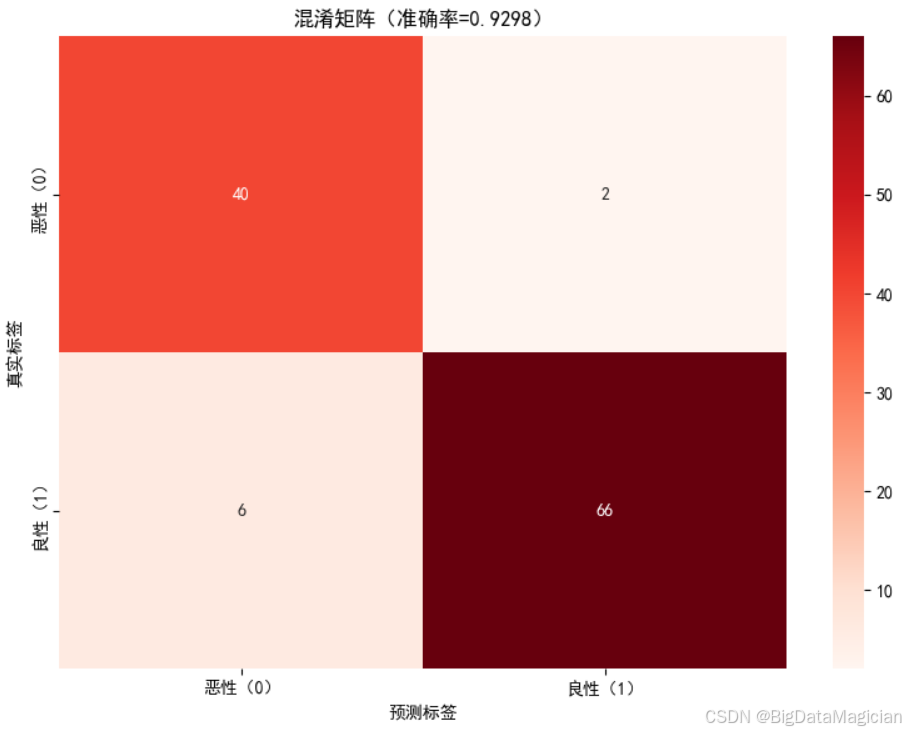
为更直观地呈现模型在测试集上的分类结果分布,将混淆矩阵以热力图形式可视化,通过颜色深浅与数值标注结合的方式,清晰展示不同类别样本的真实与预测标签对应关系。
可视化过程中,采用红色系配色(颜色越深代表样本数量越多),并在热力图单元格中标注对应样本数,同时在坐标轴与标题中明确标签含义及模型整体准确率,增强图表的可读性。
python
import seaborn as sns
import matplotlib.pyplot as plt
# 绘制混淆矩阵热力图
plt.figure(figsize=(8, 6))
sns.heatmap(
data=cm,
annot=True, # 显示单元格数值
fmt='d', # 数值格式为整数
cmap='Reds', # 配色方案
# xticklabels=cancer.target_names,
# yticklabels=cancer.target_names
xticklabels=['恶性(0)', '良性(1)'],
yticklabels=['恶性(0)', '良性(1)']
)
plt.xlabel('预测标签')
plt.ylabel('真实标签')
plt.title('混淆矩阵(准确率={:.4f})'.format(test_accuracy))
plt.tight_layout()
plt.savefig('混淆矩阵.png')从可视化结果可直观观察到,热力图中深色区域集中在"真实恶性-预测恶性""真实良性-预测良性"的对角线上,说明多数样本被正确分类;非对角线区域(误判样本)的颜色较浅且数值较小,对应仅2例恶性样本被误判为良性、6例良性样本被误判为恶性,与之前的量化结果一致。
该热力图通过视觉化方式清晰呈现了模型的分类表现,更便于快速识别误判的类别与规模,为后续模型优化提供了直观参考。
6.2.2 ROC曲线与AUC值可视化
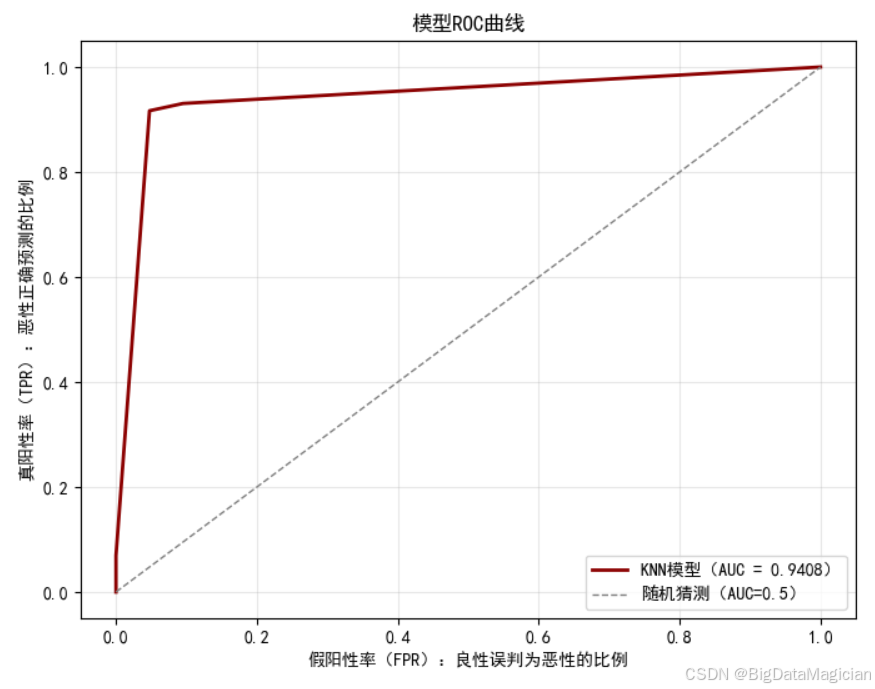
ROC曲线是评估二分类模型区分能力的核心可视化工具,通过横轴"假阳性率(FPR,即良性样本被误判为恶性的比例)"、纵轴"真阳性率(TPR,即恶性样本被正确识别的比例)"的变化关系,直观呈现模型在不同阈值下的性能表现;而AUC值(ROC曲线下的面积)则是对曲线的量化总结,值越接近1代表模型区分能力越强。
本次可视化中,以红色曲线呈现模型的ROC变化趋势,同时绘制"随机猜测"基准线(AUC=0.5)作为参照:若模型性能与随机猜测相当,曲线会贴合基准线;若模型具备区分能力,曲线则会向图的左上角(高TPR、低FPR)靠拢。
python
from sklearn.metrics import roc_curve, roc_auc_score
# 计算ROC曲线坐标
fpr, tpr, thresholds = roc_curve(y_test, y_pred_proba_best[:, 1])
auc = roc_auc_score(y_test, y_pred_proba_best[:, 1])
# 绘制ROC曲线
plt.figure(figsize=(8, 6))
# 绘制模型ROC曲线
plt.plot(fpr, tpr, color='darkred', lw=2, label=f'KNN模型(AUC = {auc:.4f})')
# 绘制随机猜测基准线(AUC=0.5)
plt.plot([0, 1], [0, 1], color='gray', lw=1, linestyle='--', label='随机猜测(AUC=0.5)')
# 设置标签与格式
plt.xlabel('假阳性率(FPR):良性误判为恶性的比例')
plt.ylabel('真阳性率(TPR):恶性正确预测的比例')
plt.title('模型ROC曲线')
plt.legend(loc='lower right')
plt.grid(alpha=0.3)
plt.savefig('模型ROC曲线.png')从结果可见,模型ROC曲线明显偏离随机猜测基准线,整体向左上角凸起,说明在多数阈值下,模型能同时保持较高的真阳性率与较低的假阳性率;对应的AUC值达0.9408,接近1,进一步验证了模型对恶性、良性样本的强区分能力------这意味着在乳腺癌诊断场景中,该模型既能有效识别恶性肿瘤(减少漏诊),又能控制对良性样本的误判(避免过度医疗)。该可视化结果直观印证了模型的分类可靠性,是评估其临床实用价值的重要依据之一。
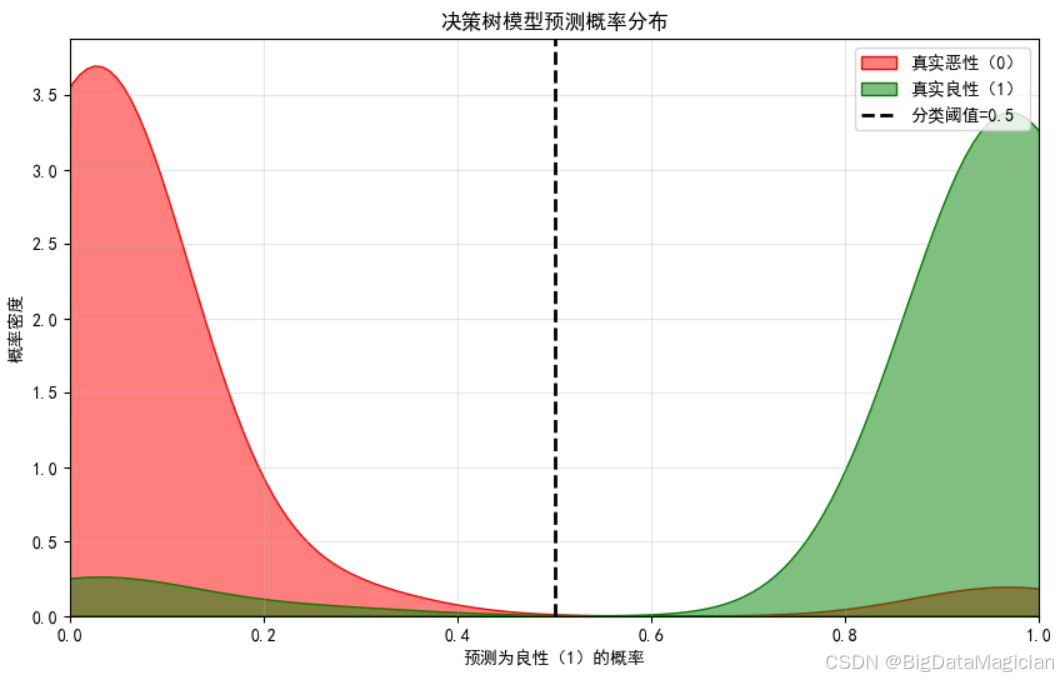
6.2.3 预测概率分布可视化
预测概率分布可视化通过核密度图,呈现不同真实类别样本的"预测为良性的概率"分布特征,可直观判断模型对两类样本的概率区分度------若两类分布重叠少,说明模型能清晰区分不同类别的样本;若重叠多,则表明模型对部分样本的概率预测模糊。
本次可视化中,分别以红色、绿色填充区域代表"真实恶性""真实良性"样本的预测概率分布,并添加默认分类阈值(0.5)辅助观察:当样本预测为良性的概率≥0.5时,模型判定为良性;反之则判定为恶性。
python
import seaborn as sns
import matplotlib.pyplot as plt
# 提取良性样本(真实标签1)和恶性样本(真实标签0)的预测良性概率
# 真实恶性→预测良性的概率
malignant_proba = test_pred_df[test_pred_df['真实标签'] == '恶性']['良性概率']
# 真实良性→预测良性的概率
benign_proba = test_pred_df[test_pred_df['真实标签'] == '良性']['良性概率']
# 绘制预测概率核密度图
plt.figure(figsize=(10, 6))
# 绘制真实恶性样本的预测概率分布
sns.kdeplot(data=malignant_proba, fill=True, color='red', alpha=0.5, label='真实恶性(0)')
# 绘制真实良性样本的预测概率分布
sns.kdeplot(data=benign_proba, fill=True, color='green', alpha=0.5, label='真实良性(1)')
# 添加分类阈值线(默认0.5)
plt.axvline(x=0.5, color='black', linestyle='--', linewidth=2, label='分类阈值=0.5')
# 设置标签与格式
plt.xlabel('预测为良性(1)的概率')
plt.ylabel('概率密度')
plt.title('决策树模型预测概率分布')
plt.xlim(0, 1) # 概率范围限定在0-1
plt.grid(alpha=0.3)
plt.legend()
plt.savefig('预测概率分布.png')从结果可见,真实恶性样本的预测良性概率集中在0-0.1区间(多数接近0),真实良性样本的预测良性概率集中在0.9~1区间(多数接近1),两类分布几乎无重叠;分类阈值(0.5)恰好处于两类分布的间隔区域,说明在默认阈值下,模型能通过概率值明确区分恶性与良性样本,仅极少数样本的概率会接近阈值导致误判。
该图从概率维度印证了模型的分类可靠性,两类样本的预测概率分布边界清晰,进一步说明模型对乳腺癌样本的分类决策具备明确的概率依据,而非模糊判定。
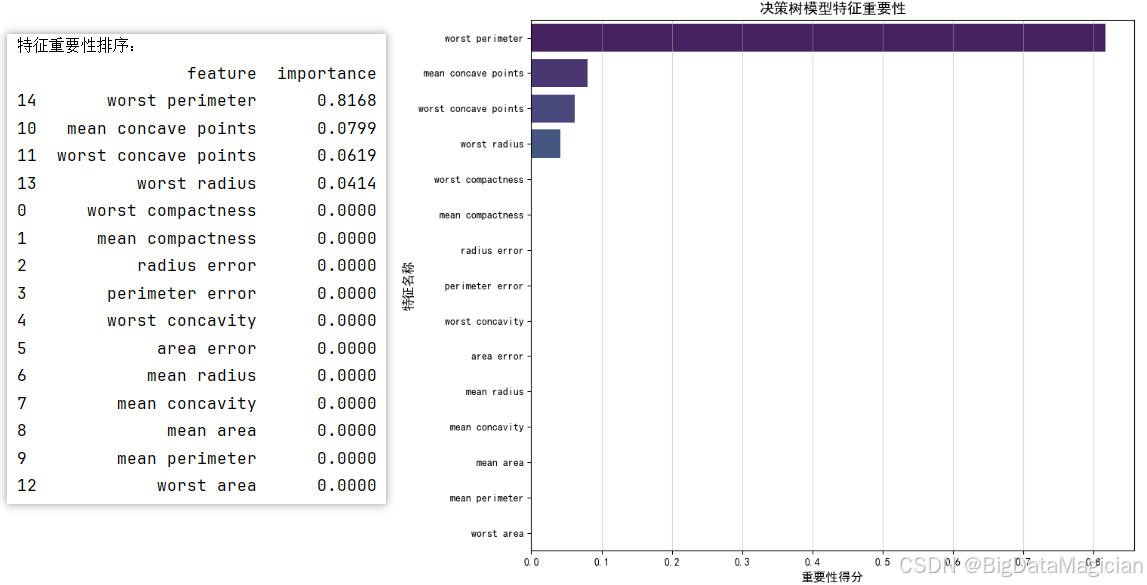
6.2.4 特征重要性可视化
特征重要性是决策树模型的核心解释性指标之一,它量化了每个输入特征对模型分类决策的贡献程度(得分越高,代表该特征在树的节点拆分过程中对纯度提升的作用越大)。通过可视化特征重要性,可明确哪些医学特征是区分乳腺癌良恶性的关键依据。
python
# 获取特征重要性
importances = dt_best.feature_importances_
feature_names = X_selected.columns # 使用你选择的关键特征列名
# 创建 DataFrame 并按重要性降序排序
feature_importance_df = pd.DataFrame({
'feature': feature_names,
'importance': importances
}).sort_values(by='importance', ascending=False)
print("\n特征重要性排序:")
print(feature_importance_df.round(4))
# 绘制水平条形图
plt.figure(figsize=(10, 8))
sns.barplot(
data=feature_importance_df,
x='importance',
y='feature',
hue='feature',
palette='viridis'
)
plt.title('决策树模型特征重要性', fontsize=14)
plt.xlabel('重要性得分', fontsize=12)
plt.ylabel('特征名称', fontsize=12)
plt.tight_layout()
plt.grid(axis='x', alpha=0.5)
plt.savefig('特征重要性.png')本次可视化将特征按重要性得分降序排列,以水平条形图展示各特征的贡献程度。从结果可见:
- 核心特征高度集中:仅"worst perimeter(最差周长)""mean concave points(平均凹点)""worst concave points(最差凹点)""worst radius(最差半径)"4个特征具有非零重要性,其中"worst perimeter"的重要性得分(0.8168)远超其他特征,是模型分类决策的主导依据;
- 多数特征无贡献:其余特征的重要性得分为0,说明这些特征在决策树的节点拆分过程中未参与模型的分类逻辑构建。
这一结果与医学认知相契合------肿瘤的周长、凹点等形态学特征是临床判断良恶性的经典指标,也验证了该决策树模型的特征选择逻辑符合实际诊疗的关注重点,同时简化了后续特征采集的成本(仅需重点关注少数核心特征)。
6.2.5 决策规则可视化
决策树的核心优势在于其决策逻辑的可解释性,通过可视化决策规则与树结构,可将模型的分类过程转化为清晰的"条件判断流程",便于理解模型如何基于特征做出分类结论。
本次可视化同时采用文本规则 与树形结构两种形式呈现,文本规则以层级缩进的方式展示节点的拆分条件与对应类别;树形结构则通过节点(包含拆分特征、基尼系数、样本量等信息)与分支(对应条件判断结果),直观呈现分类的逐层决策过程。
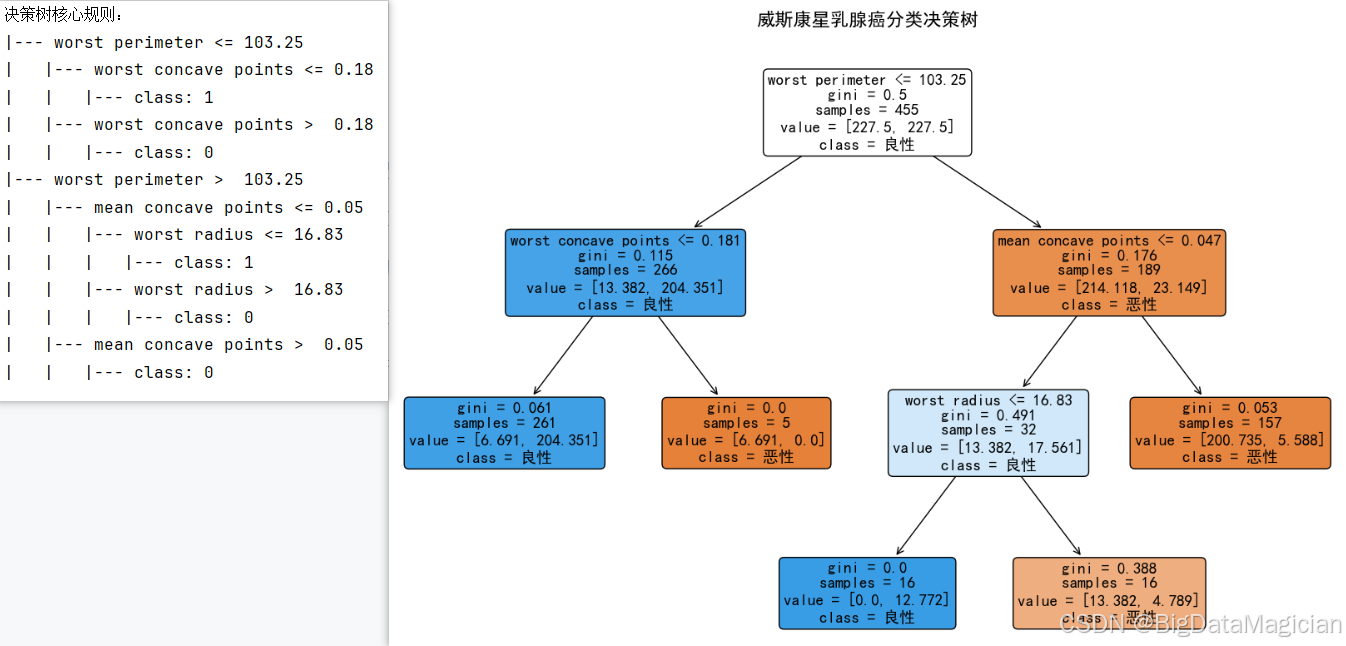
python
from sklearn.tree import export_text, plot_tree
# 导出文本格式的决策规则
tree_rules = export_text(dt_best, feature_names=list(key_features))
print("决策树核心规则:")
print(tree_rules)
# 绘制决策树结构
plt.figure(figsize=(15, 10))
plot_tree(dt_best, feature_names=key_features, class_names=['恶性', '良性'], filled=True, rounded=True)
plt.title('威斯康星乳腺癌分类决策树', fontsize=16)
plt.savefig('决策树结构.png')从结果可见,决策树的核心分类逻辑围绕核心特征展开:
- 第一层拆分:以"worst perimeter(最差周长)≤103.25"为条件,将样本分为两大分支;
- 后续层级:基于"worst concave points(最差凹点)""mean concave points(平均凹点)""worst radius(最差半径)"等核心特征进一步细分,最终输出"恶性"或"良性"的类别结论。
这种分层判断的逻辑与临床诊断的"逐步排查"思路高度契合,模型先通过最关键的特征(最差周长)做初步区分,再结合次要核心特征细化判断,既保证了分类的准确性,又让决策过程透明、可追溯------这在医疗场景中尤为重要,能帮助医生理解模型结论的依据,提升对模型的信任度。