在单细胞测序分析中,轨迹分析作为一种重要的方法,能够帮助我们深入理解细胞发育过程、分化路径以及细胞状态的动态变化。通过轨迹分析,我们可以构建细胞的时间序列,揭示细胞在特定生物学过程中如何从一个状态过渡到另一个状态。它不仅在研究发育生物学、癌症进程等方面展现出巨大潜力,还为药物开发和疾病治疗提供了新的思路。
Slingshot 推断细胞轨迹
Slingshot 是一种常用于单细胞数据分析的轨迹推断工具,它通过一种基于最小生成树(MST,Minimum Spanning Tree)的方法,构建细胞的发育轨迹。Slingshot 能够有效地处理非线性的细胞发育过程,推断细胞的分化路径,识别多个分化轨迹之间的关系,并通过视觉化方式帮助研究者理解复杂的细胞状态转变。
library(Seurat)
library(tidyverse)
load(file = "data/GSE45719/mm_seob.rdata")
mm_seob <- UpdateSeuratObject(mm_seob)
mm_seob <- SCTransform(mm_seob) %>%
RunPCA(npcs = 10) %>%
RunUMAP(dims = 1:10)
# 转换为SingleCellExperiment
sce <- as.SingleCellExperiment(mm_seob)
library(slingshot)
# 运行 Slingshot 进行轨迹推断
sce <- slingshot(sce, clusterLabels = 'cell_type',
reducedDim = 'PCA', #可替换为UMAP
start.clus = c("zy"), end.clus = "lateblast")
# 分析结果可以用 as.SlingshotDataSet 提取
sds <- SlingshotDataSet(sce)
# 提取轨迹信息
curves_df <- sds@curves[["Lineage1"]][["s"]][,c(1,2)]
colnames(curves_df) <- c("Dim1", "Dim2")
# 可视化
DimPlot(mm_seob,
reduction = "pca",
group.by = "cell_type"
) +
geom_path(data = curves_df, aes(x = Dim1, y = Dim2), group = 1, color = "black", linewidth = 1)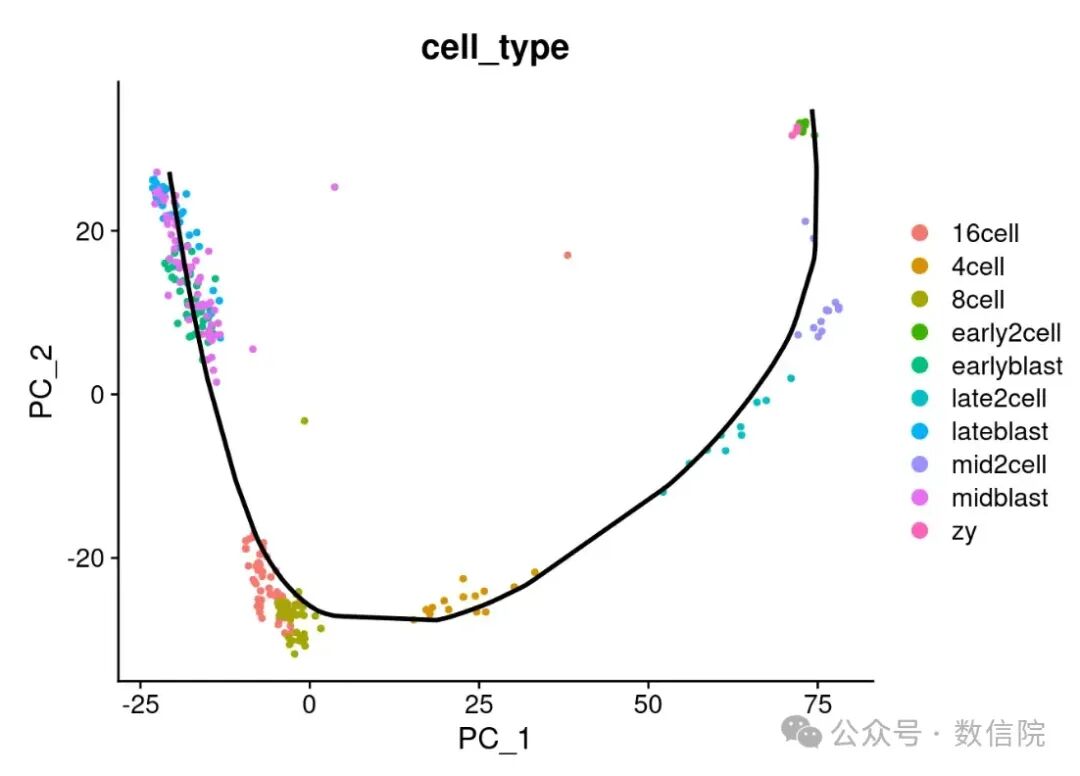
Monocle3推断细胞轨迹
Monocle3通过构建细胞轨迹模型,帮助研究者揭示细胞在生物学过程中如何从一个状态转变到另一个状态。Monocle3的核心优势在于它能够处理复杂的非线性轨迹,提供更准确的轨迹推断和细胞状态排序。
library(Seurat)
library(tidyverse)
dynrds <- readRDS(file = "data/NKT-differentiation_engel.rds")
counts <- t(dynrds[["counts"]])
cell_info <- str_remove(dynrds[["cell_info"]]$milestone_id, "vα14 inkt thymocyte subset: ")
names(cell_info) <- dynrds[["cell_info"]]$cell_id
NTK_seob <- CreateSeuratObject(counts = counts,
min.cells = 3,
min.features = 200)
NTK_seob$cell_type = cell_info[colnames(counts)]
NTK_seob <- SCTransform(NTK_seob) %>%
RunPCA(npcs = 10) %>%
RunUMAP(dims = 1:10)
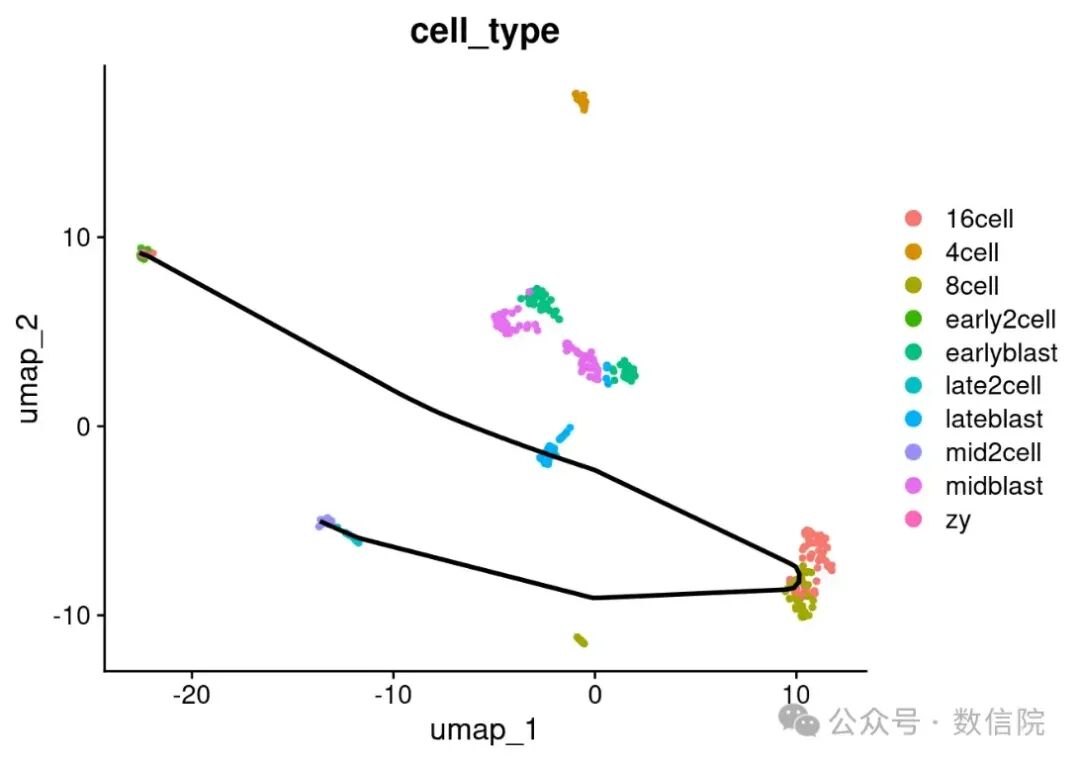
DimPlot(
NTK_seob,
reduction = "umap",
group.by = "cell_type",
label = T
) Vα14 iNKT 胸腺细胞(Vα14 invariant Natural Killer T cells, iNKT)是一种特殊的T细胞亚型,具有自然杀伤细胞(NK细胞)和T细胞的双重特性。它们通过表达特定的恒定T细胞受体(TCR),识别CD1d分子呈递的脂质抗原。Vα14 iNKT细胞在免疫反应中具有重要作用,可以快速分泌多种细胞因子,调节先天性和适应性免疫反应。胸腺中的iNKT细胞是其发育过程的一个阶段,随后会分化为不同的亚型,如NKT1、NKT2和NKT17。
library(SeuratWrappers)
library(monocle3)
# 将 Seurat 对象 seob_subset 转换为 Monocle 3 的 cell_data_set 对象
cds <- as.cell_data_set(NTK_seob,
assay = "SCT",
reductions = c("pca", "umap"),
default.reduction = "umap"
)
# 添加基因信息
rowData(cds) <- data.frame(
row.names = rownames(cds),
gene_short_name = rownames(cds)
)
# 聚类信息无法完整转移过来,使用 cluster_cells 重新聚类
cds <- cluster_cells(cds, reduction_method = "UMAP", resolution = 0.3)
# 构建细胞轨迹图
cds <- learn_graph(cds, use_partition = F, close_loop = T)
# a helper function to identify the root principal points:
get_earliest_principal_node <- function(cds, cell_type){
cell_ids <- which(colData(cds)[, "cell_type"] == cell_type)
closest_vertex <-
cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <-
igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names
(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
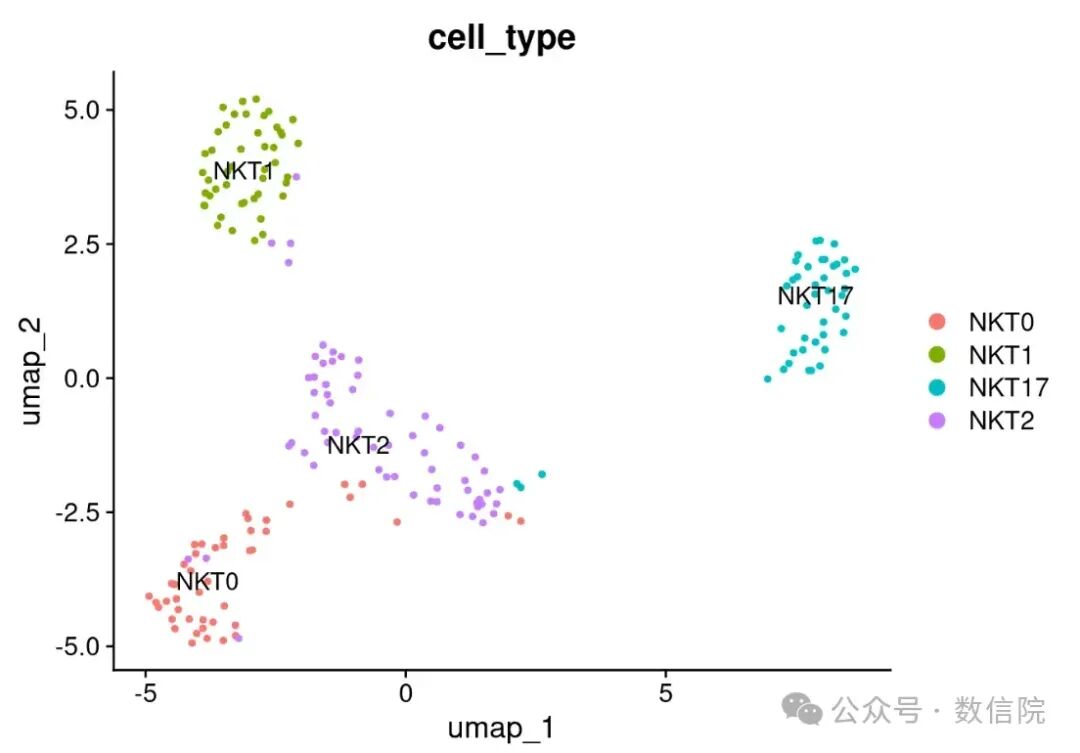
cds <- order_cells(cds,
root_pr_nodes = get_earliest_principal_node(cds, "NKT0"))
# 可视化细胞轨迹图,按聚类着色
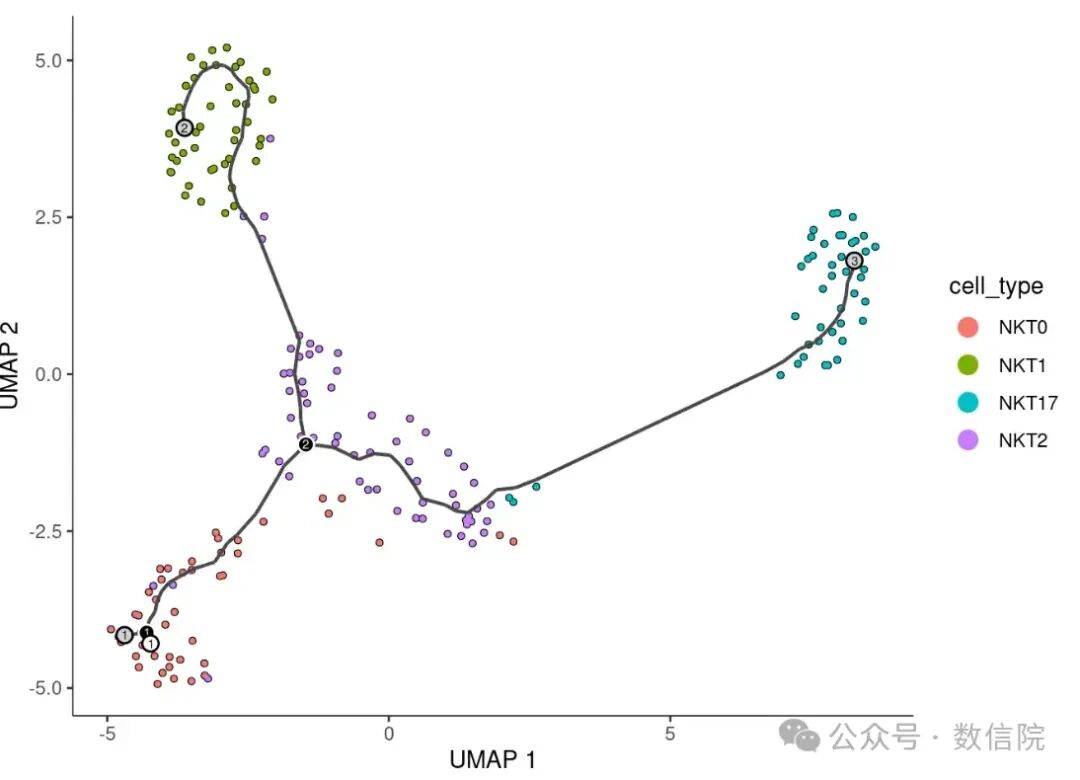
plot_cells(cds,
reduction_method = "UMAP",
#color_cells_by = "pseudotime",
color_cells_by = "cell_type",
cell_size = 0.8,
show_trajectory_graph = T, # 显示细胞轨迹
trajectory_graph_color = "grey28", # 轨迹颜色
trajectory_graph_segment_size = 0.75, # 轨迹线宽
label_cell_groups = F, # 是否在图中显示 label, F 则在图例中
label_branch_points = T, # 是否显示节点编号
label_leaves = T # 是否显示分支编号
) 
# 3D 可视化
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d, use_partition = F, close_loop = T)
cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds, "NKT0"))
cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by="cell_type")
# 随时间变化的基因
cds_pr_test_res <- graph_test(cds,
neighbor_graph="principal_graph",
cores=4)
cds_pr_test_res <- arrange(cds_pr_test_res,
desc(morans_test_statistic))
head(cds_pr_test_res)status:表示测试的状态,如果是 "OK",说明测试成功完成,没有出现错误。
p_value:这个值表示每个基因在轨迹上的空间表达关联性的显著性水平。p 值越小,说明基因在轨迹上的表达模式越显著。通常,p 值小于 0.05 被认为是显著的,但这里有多个测试,因此建议使用 q 值来进行多重比较校正。
morans_test_statistic:这个值是基于莫兰指数(Moran's I)计算的测试统计量,用于衡量基因表达值的空间自相关性。较高的统计量意味着在轨迹上基因表达的显著变化。
morans_I:莫兰指数(Moran's I)是一种衡量空间自相关性的统计量。它的值在 -1 到 1 之间:
-
正值(接近于 1)表示正空间自相关,即在轨迹上相近的细胞具有相似的基因表达模式。
-
负值(接近于 -1)表示负空间自相关,即在轨迹上相近的细胞具有相反的基因表达模式。
-
接近 0 的值表示没有明显的空间自相关。
q_value:这是对 p 值进行多重假设检验校正后的值(如 Benjamini-Hochberg 方法),用于控制假发现率(FDR)。q 值越小,说明基因在轨迹上的显著变化越可靠。通常,q 值小于 0.05 被认为是显著的。
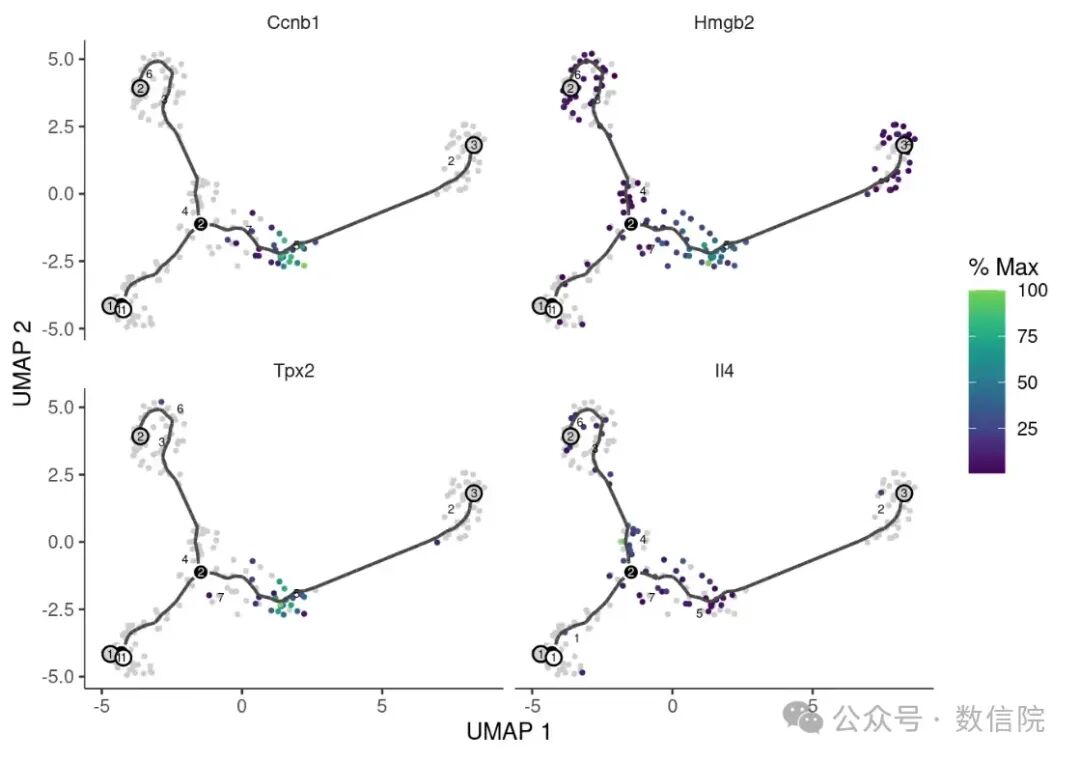
key_genes <- c("Ccnb1", "Hmgb2", "Tpx2", "Il4")
plot_cells(cds,
genes = key_genes,
cell_size = 0.8)