介绍
基于CellChat v2.20
在单细胞转录组数据中,每个细胞并不是孤立存在的,它们会不断通过分泌信号分子、表达受体等方式进行"交流"。这些交流就像细胞之间的"语言",决定了组织如何维持稳态、免疫细胞如何启动反应、发育过程中细胞如何分化成不同谱系。
细胞通讯分析(Cell--cell communication analysis) 就是利用基因表达数据来研究不同细胞类型之间如何互相"对话"。其核心思想是:
如果某一类细胞高表达某个配体(ligand) ,而另一类细胞表达其对应受体(receptor),那么两者之间可能存在潜在的信号传递。
细胞通讯分析主要包含以下内容:
- 配体--受体互作推断:识别哪些细胞在"发送信号",哪些细胞在"接收信号"。
- 信号通路活性分析:探索哪些经典通路(如 TGF-β、IL-6、WNT)在细胞之间被激活。
- 细胞互作网络构建:绘制细胞之间的通讯网络,寻找关键的调控节点。
- 通讯强度定量:评估不同细胞对之间的通讯强弱,揭示哪些细胞在组织中更"活跃"。
- 功能与生物学意义解读:结合富集分析,挖掘细胞通讯背后的潜在功能与调控机制。
目前常用的工具包括 CellChat、CellPhoneDB、iTALK、NicheNet 等,它们基于配体-受体数据库和单细胞表达矩阵,帮助研究者从复杂的数据中筛选真正具有生物学意义的细胞互作模式。
通过细胞通讯分析,研究者可以从整体视角理解细胞如何相互协调,在疾病、发育或免疫反应中承担什么角色,从而为后续实验验证和机制研究提供重要线索。
CellChat 相较于其他工具,如 SingleCellSignalR、iTALK 和 NicheNet,具有显著优势,它考虑了许多受体作为多亚基复合物存在的情况。并且可视化效果也更好。本篇文章就以cellchat为例进行细胞通讯分析.
方法分类
单细胞转录组中CellChat官网有三种方式
单一数据集
就是一个样本进行分析(不过现在大多都是多个样本合并进行分析,但是是建议单个样本分析或者同类样本合并分析),例如本次7个甲状腺组织,一个癌旁,六个PTC,就可以将6个PTC合并作为一个样本和癌旁比对;
多样本分析
是对比不同样本之间互作的方式和强度(最常见);
多个不同细胞组成的细胞集
解决了一个很现实的问题,首先我们要了解,想比较两个样本之间的细胞通讯强度、模式、网络结构,就必须保证:
两个样本的细胞类型分类(labels)一致,否则无法直接对比。
例如一个数据集A有11种细胞,另一个数据集B在11种细胞的基础上多两种细胞,CellChat 提供的 liftCellChat() 就是专门解决这个问题的,给数据集 A 创建一个空的占位标签(no cells),从而保证两组标签一致。
流程介绍
本次介绍单一数据集分析流程;以7个样本作为一个样本整体分析
CellChat 的输入数据结构非常固定,只需要 两个核心数据**,细胞表达矩阵(标准化之后)和细胞 metadata(带有 cell type 信息)**
对于单个样本细胞注释信息不需要太过详细,CellChat 默认要求: 每个 cluster 至少 **10 个细胞,**单个样本的细胞数量无法满足很详细的注释,但是对于多个样本的分析中,更加推荐详细的注释能大幅提升通讯解析的生物学解释力。
例如
粗注释 "T cell" 的通信图
CellChat 会显示: T cell → Myeloid; T cell → Fibroblast;T cell → Epithelial 只能看到整体趋势。
精细注释 T cell 后 就可以得到:**Treg → Myeloid(过度抑制型通讯,例如 TGFB);CD8 exhausted → Tumor(PD-L1/PD-1 通路);CD4 Tfh → B cell(CD40/CD40L 信号增强);CD8 TRM → Epithelial(IL-7, IL-15 通讯)**这些才是免疫治疗、肿瘤微环境研究的关键发现。
数据准备
R
library(Seurat)
library(CellChat)
# 读取之前的数据
scRNA <- readRDS("D:/datat/新建文件夹/GSE191288_RAW/scRNA.rds")
T_sce <- readRDS("D:/datat/新建文件夹/GSE191288_RAW/T_sce.rds")
B_sce <- readRDS("D:/datat/新建文件夹/GSE191288_RAW/B_sce.rds")
# 子集注释回写母对象
phe <- scRNA@meta.data
phe_T <- T_sce@meta.data
phe_B <- B_sce@meta.data
# 如有因子,先转换为字符
phe$celltype <- as.character(phe$celltype)
phe_T$celltype.2 <- as.character(phe_T$celltype.2)
phe_B$celltype <- as.character(phe_B$celltype)
# 找到匹配的行
common_T <- intersect(rownames(phe), rownames(phe_T))
common_B <- intersect(rownames(phe), rownames(phe_B))
# 回写细胞类型
phe[common_T, "celltype"] <- phe_T[common_T, "celltype.2"]
phe[common_B, "celltype"] <- phe_B[common_B, "celltype"]
scRNA@meta.data <- phe
## 细胞的基因表达数据
data.input <- GetAssayData(scRNA, slot = 'data') # normalized data matrix
## metadata文件
meta <- scRNA@meta.data[,c("orig.ident","celltype")]
# 根据研究情况进行细胞排序
celltype_order <- c(
"CD 4 naive",
"CD 4 TCM",
"CD 4 Tem/Th1-like",
"CD 4 Tfh",
"CD 4 TRM",
"Treg",
"CD 8 Exhausted",
"CD 8 TEM",
"CD 8 Temra/TeFF",
"CD 8 TRM",
"gy T",
"NKT",
"NK",
"Naive B",
"Activated B",
"Memory B cells",
"Resting Memory B cells",
"Plasma cells",
"Endothelial",
"Epithelial Cells",
"Fibroblasts Cells",
"Myeloid Cells "
)
# 对 meta 和 data.input 进行排序
identical(rownames(meta), colnames(data.input))
meta$celltype <- factor(meta$celltype ,levels = celltype_order)
ordered_indices <- order(meta$celltype)
meta <- meta[ordered_indices, ]
data.input <- data.input[, ordered_indices]
identical(rownames(meta),colnames(data.input))创建cellchat对象
R
cellchat <- createCellChat(object = data.input, meta = meta, group.by = "celltype")
# 添加细胞信息
cellchat <- addMeta(cellchat, meta = meta)
cellchat <- setIdent(cellchat, ident.use = "celltype")
levels(cellchat@idents)
groupSize <- as.numeric(table(cellchat@idents))设置配体受体交互数据库
CellChat 的核心原理是通过 配体-受体数据库(CellChatDB) 推断细胞间通讯。
CellChatDB 包含:
- Secreted Signaling(分泌型信号,如 chemokine、cytokine)
- Cell--cell Contact(细胞接触)
- ECM--Receptor(基质-受体)
- Non-protein Signaling
- 代谢信号
- 神经突触信号
- 旧版(CellChatDB v1)
- 新版(CellChatDB v2)(新增可选项)
R
CellChatDB <- CellChatDB.human #人源样本
# 查看数据的分类情况与
# 具体内容showDatabaseCategory(CellChatDB)
dplyr::glimpse(CellChatDB$interaction)
#筛选数据库,只保留"Secreted Signaling"
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling")
# 如果你只想研究:细胞释放信号(cytokines、chemokines、growth factors),免疫调控,肿瘤微环境通讯
# 这是 CellChat 最典型的使用方式 推荐用于大多数免疫、肿瘤、生物学研究
# CellChatDB.use <- subsetDB(CellChatDB) 这个意思是排除非蛋白信号,官方推荐
# CellChatDB.use <- CellChatDB 这个意思是使用所有数据库,不是很推荐,因为数据很多,可视化也很杂
# 数据库写入到自己创建的cellchat中
cellchat@DB <- CellChatDB.use
预处理chellchat
R
cellchat <- subsetData(cellchat) # 只保留 配体/受体基因的表达矩阵
# 类似于找高变基因
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)计算通信概率并推断cellchat网络
R
cellchat <- computeCommunProb(cellchat, type = "triMean")
cellchat <- filterCommunication(cellchat, min.cells = 10)在信号通路级别推断细胞-细胞通信并整合的细胞通信网络
R
cellchat <- computeCommunProbPathway(cellchat)
cellchat <- aggregateNet(cellchat)可视化
所有细胞环形网络图
R
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
# 每个细胞类型之间 有多少条 ligand--receptor 互作
netVisual_circle(cellchat@net$count, vertex.weight = groupSize, weight.scale = T, label.edge= F, title.name = "Number of interactions")
# 每个细胞类型之间 这些互作的整体强度 / 权重
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize, weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")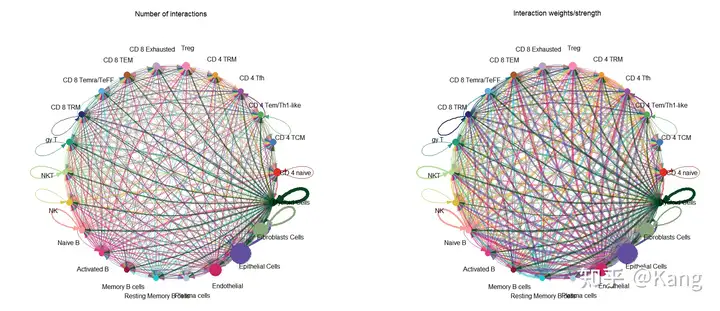
这张图展示: 某细胞类型 → 另一细胞类型 有多少条 ligand--receptor 信号通讯 / 所有 LR 对的信号强度总和
图中元素含义:
- 节点大小(vertex.weight = groupSize) = 该细胞类型的细胞数量
- 连线粗细 = 两细胞类型之间 LR 对的数量 / Signal strength(第二张)
- 线的多少 = 细胞类型之间通讯网络的复杂程度
建议两张图一起看;因为: 数量多 ≠ 强度强 强度强 ≠ 数量多
通过两张图对比,你可以判断:哪些细胞"频繁交流"(count 高);哪些细胞"信号强烈"(weight 高)
结果看着很杂乱,这种情况在一定程度上是预期之中的,因为本次分析中我们将 7 个样本 (其中还包含 1 个癌旁组织 )合并在一起进行 CellChat 推断。图像杂乱本身并不代表分析错误,而是 样本间异质性(heterogeneity)过大 所导致的正常现象。所以跟开头一样,更推荐大家做单独样本(或者同类样本合并)分析。
每个细胞环形网络图
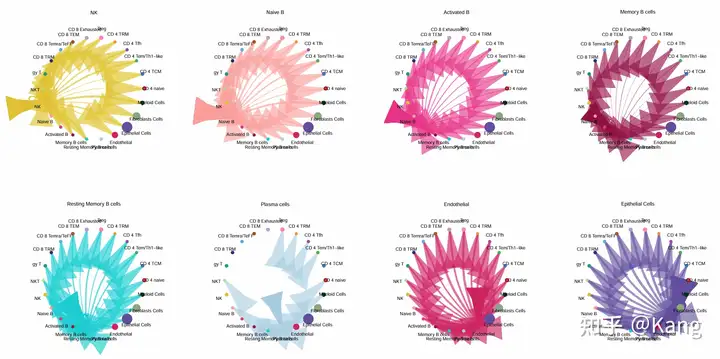
细胞通信网络复杂,我们可以检查每个细胞组发送的信号。在这里,我们还控制参数edge.weight.max,以便我们可以比较不同网络之间的边缘权重。就是把上一张图拆开绘制
R
mat <- cellchat@net$weight
par(mfrow = c(3,4), xpd=TRUE)
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(mat2, vertex.weight = groupSize, weight.scale = T, edge.weight.max = max(mat), title.name = rownames(mat)[i])
}
自定义细胞通路可视化
在推断出细胞间通讯网络之后,CellChat 提供了多种功能用于进一步的数据探索、分析和可视化。具体包括
CellChat 提供多种方法用于展示细胞间通讯网络,包括:层级图(hierarchical plot);环形图(circle plot);弦图(Chord diagram);
这些可视化方式能够从不同角度呈现细胞群之间的信号互作关系。
cellchat@netP$pathways显示所有重要通信的信号通路
R
cellchat@netP$pathways
[1] "MIF" "GALECTIN" "CXCL" "CypA" "MK" "PARs" "TGFb" "VISFATIN" "TNF" "PTN"
[11] "VEGF" "IFN-II" "CD70" "IL16" "ANNEXIN" "BTLA" "EGF" "ANGPTL" "RESISTIN" "CD137"
[21] "TWEAK" "CD40" "LIGHT" "GDF" "BAFF" "PDGF" "IL2" "GRN" "ANGPT" "GAS"
[31] "PLAU" "COMPLEMENT" "FASLG" "OSM" "PROS" "CSF" "TRAIL" "CALCR" "CCL" "APRIL"
[41] "SEMA3" "HGF" "NRG" "CX3C" "FGF" "BAG" "BMP" "SLIT" "PERIOSTIN" 我们以输入一个信号通路("CXCL")为例
R
pathways.show <- c("CXCL")
## 层次图(Hierarchy plot)
vertex.receiver = seq(1,4) #哪些细胞群体属于"接收端(receiver)
netVisual_aggregate(cellchat, signaling = pathways.show, vertex.receiver = vertex.receiver,layout = "hierarchy")
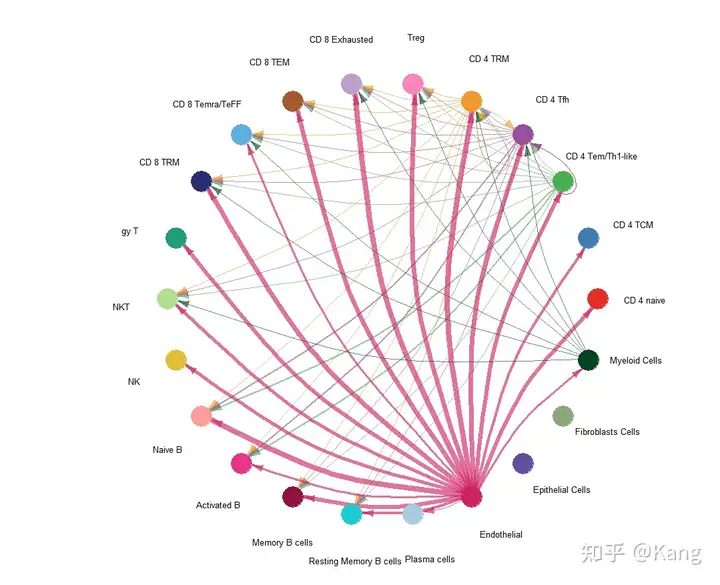
## 环形图(circle plot)
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")
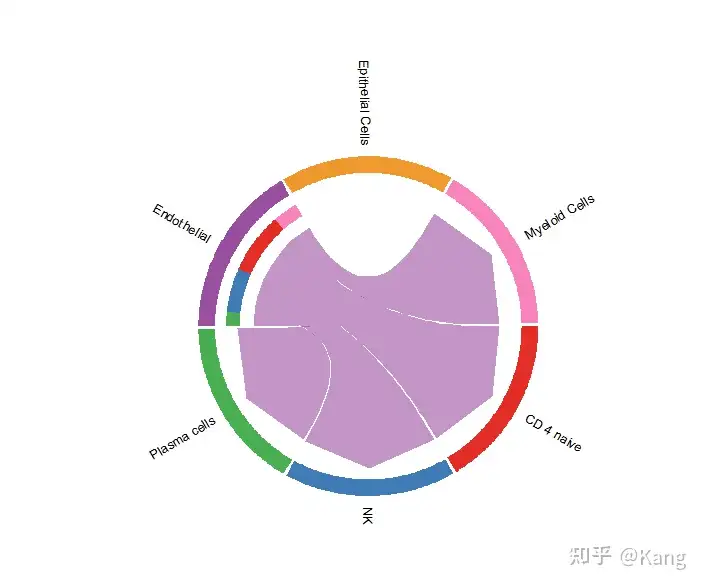
##弦图(Chord diagram)
#由于弦图细胞类型过多会报错,我们仅仅提取6类细胞绘制
cellchat.use <- subsetCellChat(cellchat,
idents.use = c("CD 4 naive", "Epithelial Cells","NK","Endothelial","Plasma cells","Myeloid Cells"))
netVisual_aggregate(cellchat.use, signaling = pathways.show, layout = "chord")
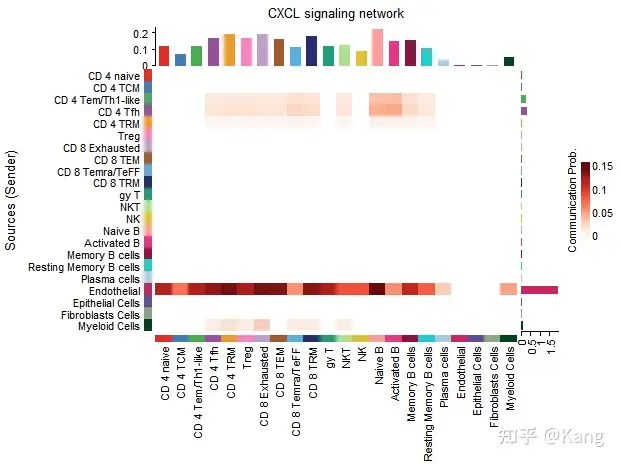
## 热图
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")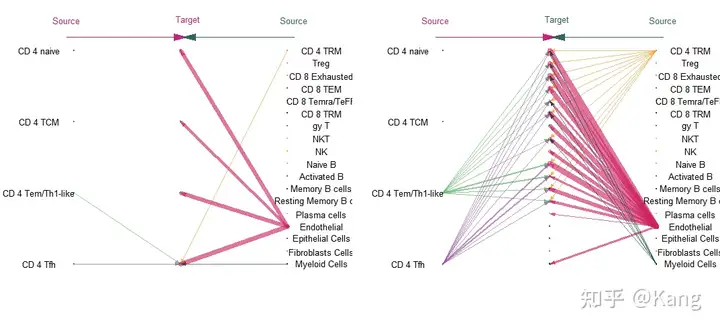
层次图(Hierarchy plot)
环形图(circle plot)
弦图(Chord diagram)
热图
计算每个配体受体对整体信号通路的贡献,并可视化由单个配体受体对调节的细胞通信
R
netAnalysis_contribution(cellchat, signaling = pathways.show)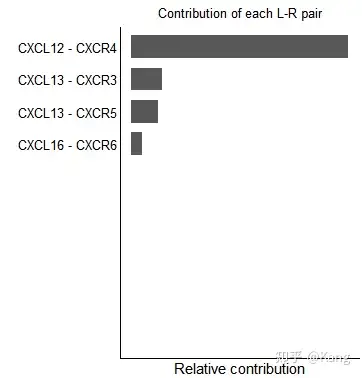
展示单个配体
可视化由单个配体受体对调节的细胞-细胞通信。我们提供一个函数extractEnrichedLR来提取给定信号通路的所有重要相互作用(L-R对)和相关信号基因。
R
pairLR.CXCL <- extractEnrichedLR(cellchat, signaling = pathways.show, geneLR.return = FALSE)
LR.show <- pairLR.CXCL[1,] # 只展示一个配体对
# Hierarchy plot
vertex.receiver = seq(1,4) # a numeric vector
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show, vertex.receiver = vertex.receiver)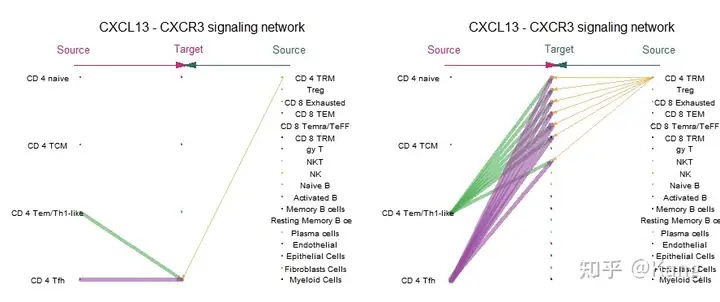
同样也可以绘制圈图;弦图
R
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show, layout = "circle")
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show, layout = "chord")自动保存所有推断网络的模块以进行快速探索
在实际使用中,如果自己研究没有目标通路,用户可以使用'for ... loop'自动保存所有推断网络快速探索使用,然后从输出文件中挑选合适的通路
R
pathways.show.all <- cellchat@netP$pathways
# check the order of cell identity to set suitable vertex.receiver
levels(cellchat@idents)
vertex.receiver = seq(1,4)
for (i in 1:length(pathways.show.all)) {
# Visualize communication network associated with both signaling pathway and individual L-R pairs
netVisual(cellchat, signaling = pathways.show.all[i], vertex.receiver = vertex.receiver, layout = "hierarchy")
# Compute and visualize the contribution of each ligand-receptor pair to the overall signaling pathway
gg <- netAnalysis_contribution(cellchat, signaling = pathways.show.all[i])
ggsave(filename=paste0(pathways.show.all[i], "_L-R_contribution.pdf"), plot=gg, width = 3, height = 2, units = 'in', dpi = 300)
}通讯网络推断
多个配体受体或信号通路调节的细胞通信
我们还可以使用netVisual_bubble显示从某些细胞组到其他细胞组的所有重要相互作用(L-R对)。
R
netVisual_bubble(cellchat, sources.use = 4, targets.use = c(5:15), remove.isolate = FALSE)
# sources.use = 4 表示选择第 4 个细胞群,表示从细胞群4发出信号,示例数据为CD 4 Tfh
# targets.use = c(5:15) 表示选择第 5 到第 15 个细胞群作为目标细胞,接受信号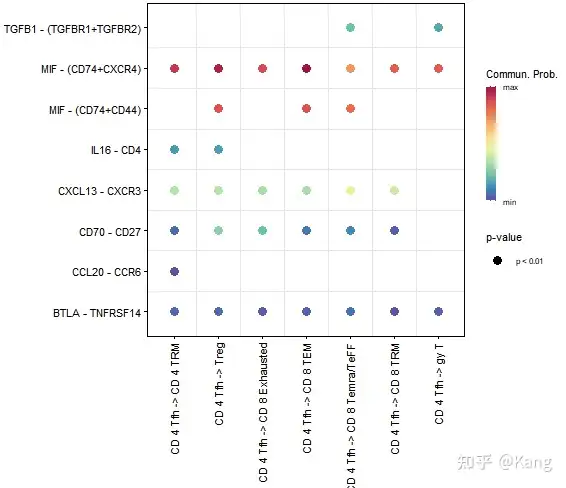
- 横轴 (x-axis) :代表目标细胞群体(接收者),例如图中列出的
CD 4 Tfh → CD 4 TRM、CD 4 Tfh → CD 8 TEM等。 - 纵轴 (y-axis) :代表信号源,也就是提供信号的细胞群体,比如图中列出的
TGFβ1 (TGFBR1+TGFBR2)、MIF (CD74+CXCR4)等。 - 气泡的大小:气泡的直径或大小通常表示某个信号的重要性,通常与信号强度或表达水平成比例。例如,较大的气泡表示信号强度较高。
- 气泡的颜色 :
- 根据
p-value的显著性,气泡的颜色代表不同的显著性水平。通常,颜色的深浅程度反映了p-value的大小,例如:- 深蓝色/紫色气泡表示
p < 0.01,这表明结果显著。 - 较浅的颜色可能表示较高的
p-value,说明结果不那么显著。
- 深蓝色/紫色气泡表示
- 根据
R
# 可以是绘制想要的通路
netVisual_bubble(cellchat, sources.use = 4, targets.use = c(5:11), signaling = c("CCL","CXCL"), remove.isolate = FALSE)
## 绘制弦图
netVisual_chord_gene(cellchat, sources.use = 4, targets.use = c(5:11), lab.cex = 0.5,legend.pos.y = 30)
使用小提琴/点图绘制信号基因表达分布
R
plotGeneExpression(cellchat, signaling = "CXCL")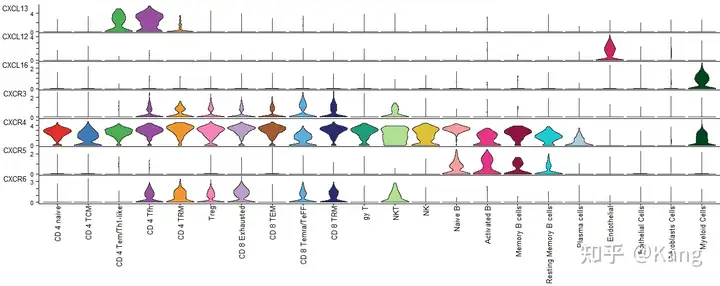
细胞通信网络系统分析
细胞通信网络系统分析是通过量化细胞之间复杂的信号传递过程,揭示细胞在组织和器官中的协同作用。CellChat 提供了一种先进的方法,利用图形理论、模式识别和多重学习来对这些细胞间的信号通路进行深入分析,从而更好地理解它们的功能和作用。以下是该分析方法的简要科普总结:
细胞通信网络分析的核心内容:
- 信号源和目标的识别:CellChat 通过集中度分析来确定信号网络中的主要信号源(发送者)和目标(接收者),这有助于识别在细胞通信中最重要的细胞类型。
- 模式识别与预测:通过模式识别方法,CellChat 能够预测特定细胞类型的关键信号通路,包括它们接收和传递信号的能力,以及不同细胞之间如何协同响应。
- 信号通路的分组:CellChat 采用相似度度量和拓扑分析,将相似的信号通路进行分组,帮助我们理解信号网络的组织结构。
- 联合多重学习:通过将多个网络的学习结果结合,CellChat 可以识别和描绘上下文特定的信号通路,揭示不同生物环境中细胞通信的变化。
- 信号角色识别:CellChat 能够识别不同细胞组在网络中的角色,如主要的信号发送者、接收者、调解者或影响者。这有助于我们理解细胞在特定生物学过程中的主导作用。
关键网络测量:
CellChat 通过计算每个细胞组的多个网络中心性指标,帮助我们识别细胞间通信中的关键参与者。以下是几种重要的度量方法:
- 外向度(Out-degree):衡量一个细胞群体向其他细胞群体发送信号的能力,通常用来识别信号的主要发送者。
- 内向度(In-degree):衡量一个细胞群体从其他细胞群体接收信号的能力,通常用来识别主要的接收者。
- 介于度(Betweenness centrality):衡量细胞群体在信息流动中的中介作用,揭示哪些细胞在信号网络中扮演着调解或桥梁的角色。
- 信息中心度(Information centrality):衡量细胞群体在整个网络信息流动中的重要性,显示哪些细胞对细胞通信有最大影响。
通过这些指标,CellChat 能够揭示细胞通信网络中的核心细胞群,帮助研究者理解细胞间的复杂互动及其在生理和病理过程中的作用。
计算和可视化网络中心分数
R
# 计算 CellChat 对象中细胞间通信网络的中心性度量
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP")
# 绘制细胞通信网络中不同细胞群体的信号角色图,展示每个细胞群体在特定信号通路中的角色
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 8, height = 2.5, font.size = 10)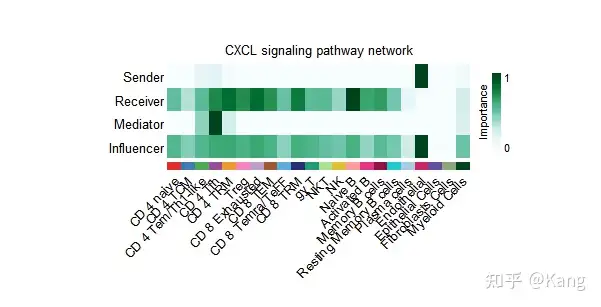
图的解读:
- 节点:图中的每个节点代表一个细胞群体(例如,T 细胞、B 细胞、上皮细胞等)。
- 边:连接节点的边表示细胞群体之间的配体-受体信号交互。边的颜色和粗细通常代表信号的强度或显著性。
- 角色分配:每个细胞群体的角色(发送者、接收者、调解者等)会在图中用不同的颜色或标记显示出来,帮助你理解在某一特定信号通路中,每个细胞群体的功能。
例如,发送者 的细胞群体会以红色或加粗的方式显示,接收者的细胞群体可能是蓝色或较细的边线。通过这些可视化结果,你可以快速识别在特定信号通路中哪些细胞是主要的信号传递者,哪些细胞是信号的主要接收者,以及哪些细胞起到中介作用。
cellchat还提供了另一种直观方法,使用散点图在 2D 空间中可视化占主导地位的发射器(源)和接收器(目标)。
R
# 对 CellChat 对象中的细胞间通信网络进行信号角色分析
gg1 <- netAnalysis_signalingRole_scatter(cellchat)
# 对指定的信号通路(在这个例子中是 CXCL 和 CCL)进行信号角色分析,并绘制散点图
gg2 <- netAnalysis_signalingRole_scatter(cellchat, signaling = c("CXCL", "CCL"))
gg1 + gg2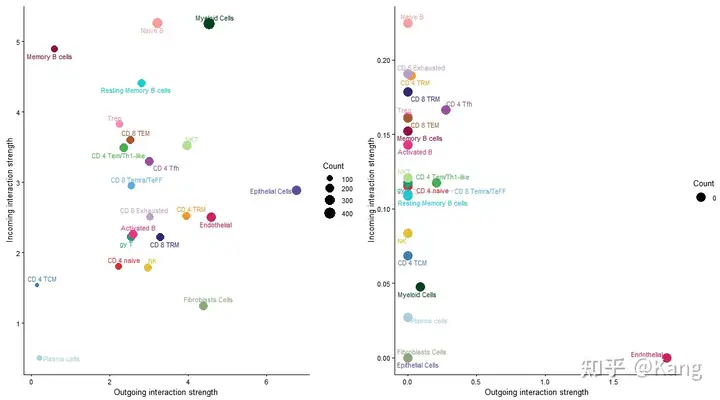
cellchat还可以回答以下问题:哪些信号对某些细胞组的传出或传入信号贡献最大。
R
ht1 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "outgoing")
ht2 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "incoming")
ht1 + ht2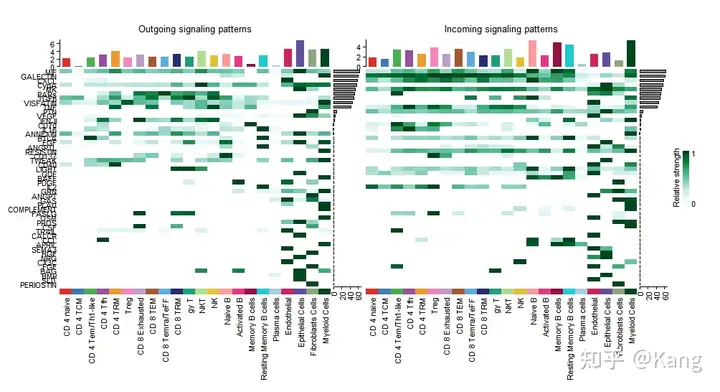
在 CellChat 中,通信模式分析旨在识别多个细胞群体和信号通路如何协调合作,共同驱动细胞间的通信。通过模式识别方法,CellChat 可以揭示全局通信模式,并提供一种方式来探究不同细胞类型和信号通路如何共同作用。以下是该分析的概述:
全局通信模式分析:
1.模式识别方法:
- CellChat 采用 非负矩阵分解(NMF)方法来识别全局通信模式,帮助确定多个细胞类型和信号通路如何协同工作。
- 默认情况下,CellChat 选择 5 种模式,但用户可以根据数据调整模式数量。
2.模式数量的选择:
- selectK 函数帮助推断最合适的模式数量,基于 Cophenetic 和 Silhouette 指标,这些指标衡量了在分层聚类共识矩阵基础上,特定模式数量的稳定性。
- 选择的模式数量通常大于 2,且应具有生物学意义。当 Cophenetic 和 Silhouette 指标值开始急剧下降时,表明已选择的模式数量较为适合。
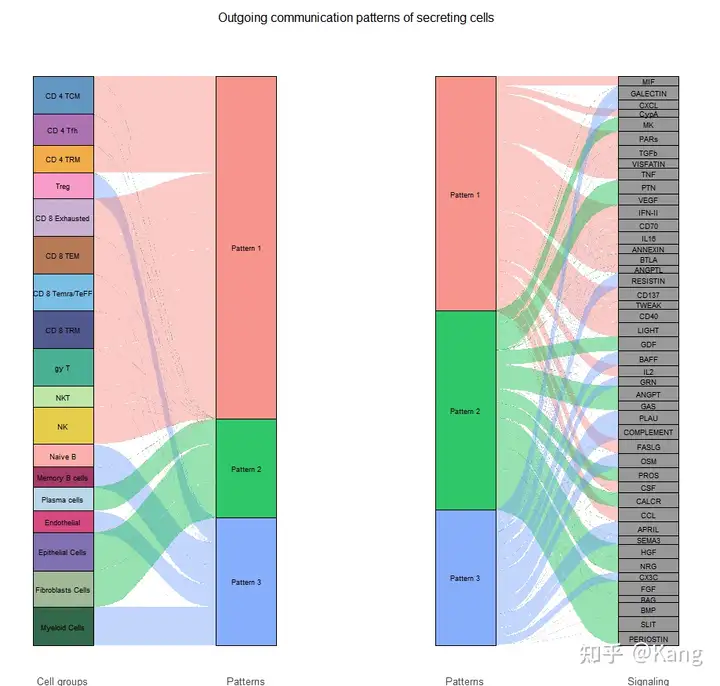
3.识别和可视化传出通信模式:
- 传出模式揭示了细胞发送信号的协调方式,显示了哪些细胞群体通过特定信号通路来传递信息。
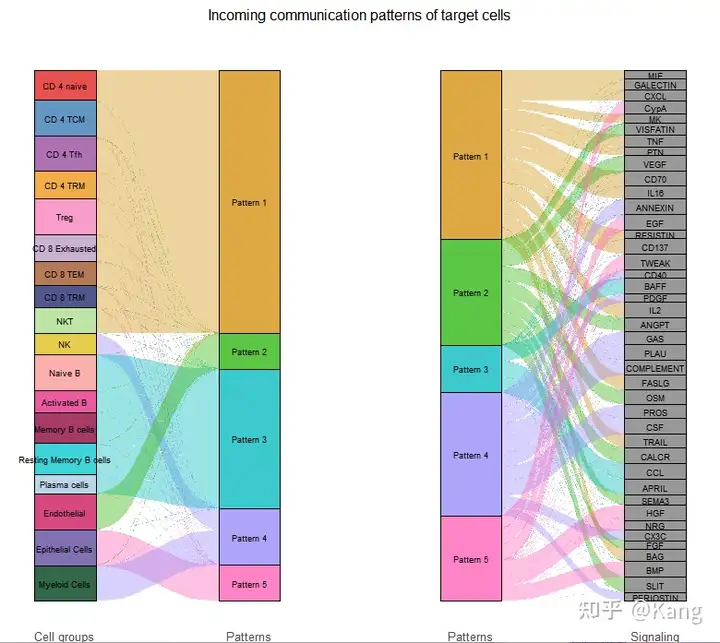
- 使用 河流图(冲积图) 可直观显示模式与细胞群体、配体受体对或信号通路之间的关系。
4.图像生成和矩阵处理:
- 为了突出与每个推断模式相关的细胞群体和信号通路,CellChat 将矩阵 W 和 H 中的元素标准化为 0,1,并对低于阈值(如 0.5)的元素进行置零。
- 这种阈值设置使得每个细胞组与特定模式之间的关系更加明确,便于可视化不同模式与细胞群体的关系。
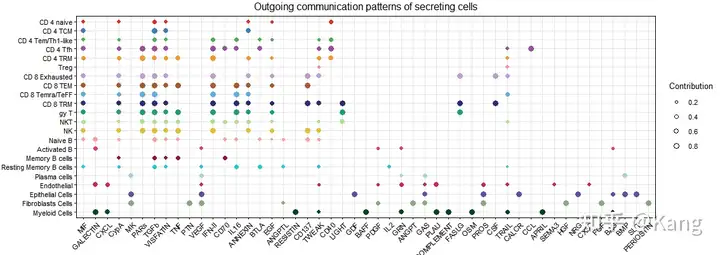
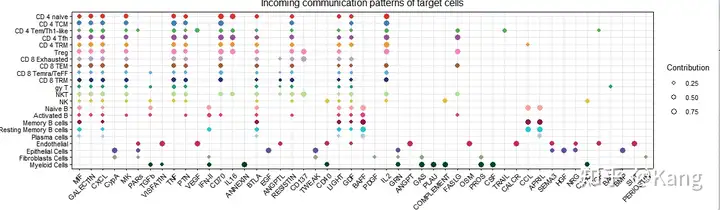
5.贡献分数计算:
- 每个细胞组和信号通路的贡献分数基于矩阵 W 和 H 的乘积计算。通过计算得分,可以构建一个点图,点的大小代表细胞群体与其丰富信号通路之间的关联强度。
6.参数调整:
-
- 用户可以调整参数 cutoff,降低阈值,以显示更多细胞组与信号通路的关联,从而获得更多的通信模式信息。
所需的包:
- CellChat:用于分析细胞间的通信网络和信号通路。
- NMF:用于进行非负矩阵分解,识别全局通信模式。
该分析方法通过识别细胞群体和信号通路之间的协调模式,帮助研究者深入了解细胞如何在不同的生物学条件下进行有效的通信。通过选择合适的模式数量和使用可视化工具(如冲积图),研究者可以更加清晰地展示不同细胞类型和信号通路之间的相互作用及其生物学意义。
传出通信模式
R
library(NMF)
library(ggalluvial)
# 推断模式的数量
selectK(cellchat, pattern = "outgoing")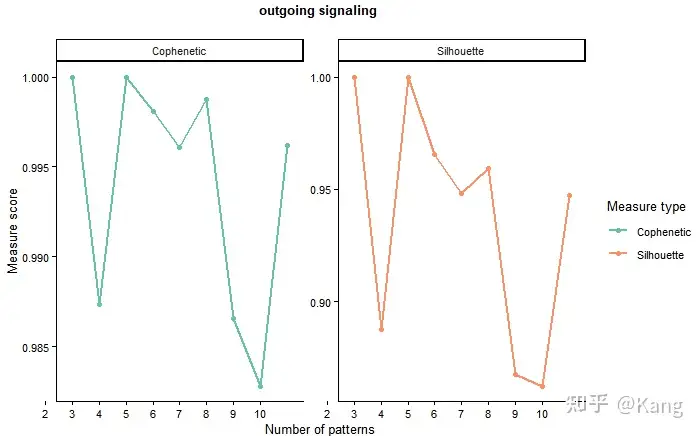
当传出模式数为 3 时,Cophenetic 和Silhouette值都开始突然下降。
R
nPatterns = 3
# 别细胞群体的 传出通信模式
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "outgoing", k = nPatterns)
# 绘制 河流图(River plot)来可视化传出通信模式
netAnalysis_river(cellchat, pattern = "outgoing")
# 绘制 点图(Dot plot)来显示传出通信模式的详细信息
netAnalysis_dot(cellchat, pattern = "outgoing")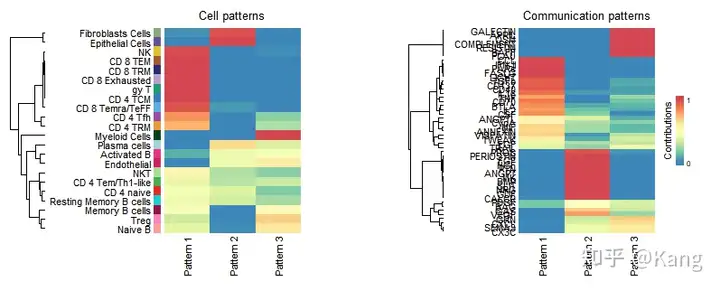
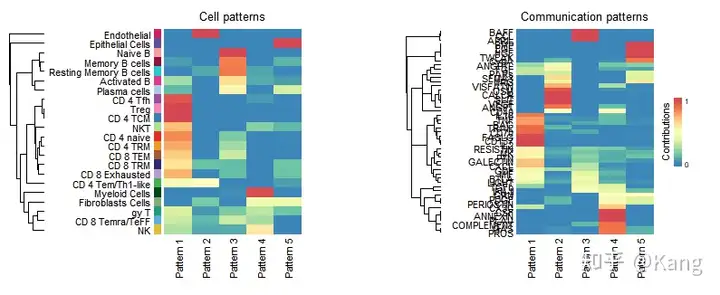
识别和可视化目标细胞的传入通信模式
方法和上面类似,传入模式显示目标细胞(即信号接收器中的细胞)如何相互协调,以及它们如何与某些信号通路协调以响应传入的信号。
R
selectK(cellchat, pattern = "incoming")
nPatterns = 5
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "incoming", k = nPatterns)
netAnalysis_river(cellchat, pattern = "incoming")
netAnalysis_dot(cellchat, pattern = "incoming")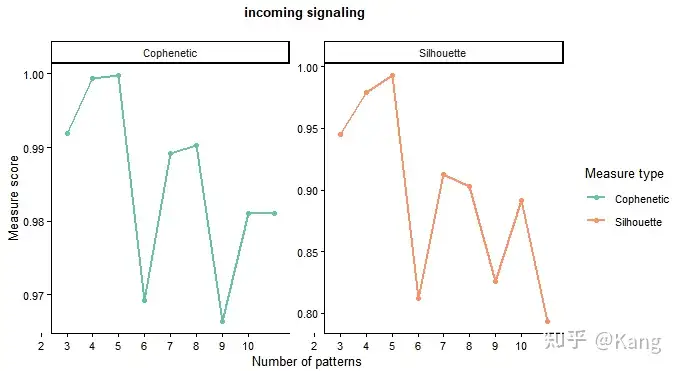
信号网络的多重和分类学习分析
此外,CellChat 能够量化所有重要信号通路之间的相似性,然后根据其CellChat 网络的相似性对其进行分组。分组可以基于功能或结构相似性进行。
功能相似性 :功能相似度高表示主要发送器和接收器相似,可解释为两个信号通路或两个配体受体对具有相似的作用。功能相似性分析要求两个数据集之间的细胞群组成相同。
结构相似性:结构相似性用于比较其信号网络结构,而不考虑发送器和接收器的相似性。
根据信号组的功能相似性识别信号组
R
# 运行时需要加载
reticulate::py_install(packages = 'umap-learn')
cellchat <- computeNetSimilarity(cellchat, type = "functional")
cellchat <- netEmbedding(cellchat, type = "functional")
cellchat <- netClustering(cellchat, type = "functional")
netVisual_embedding(cellchat, type = "functional", label.size = 3.5)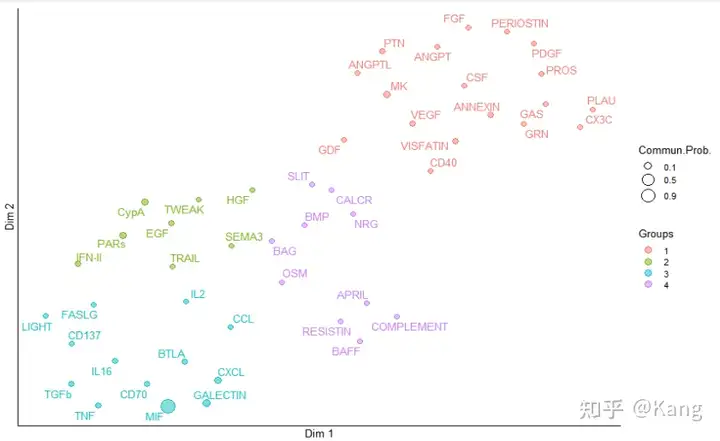
基于结构相似性识别信号组
R
cellchat <- computeNetSimilarity(cellchat, type = "structural")
cellchat <- netEmbedding(cellchat, type = "structural")
cellchat <- netClustering(cellchat, type = "structural")
netVisual_embedding(cellchat, type = "structural", label.size = 3.5)
# 分布绘制
netVisual_embeddingZoomIn(cellchat, type = "structural", nCol = 2)最后保存
R
saveRDS(cellchat, file = "cellchat.rds")