
撰文|Will
WGCNA 一句话:把「一堆基因」按「表达相似性」打包成「模块」,再挑跟「表型」最亲的模块,挖里面的核心基因。
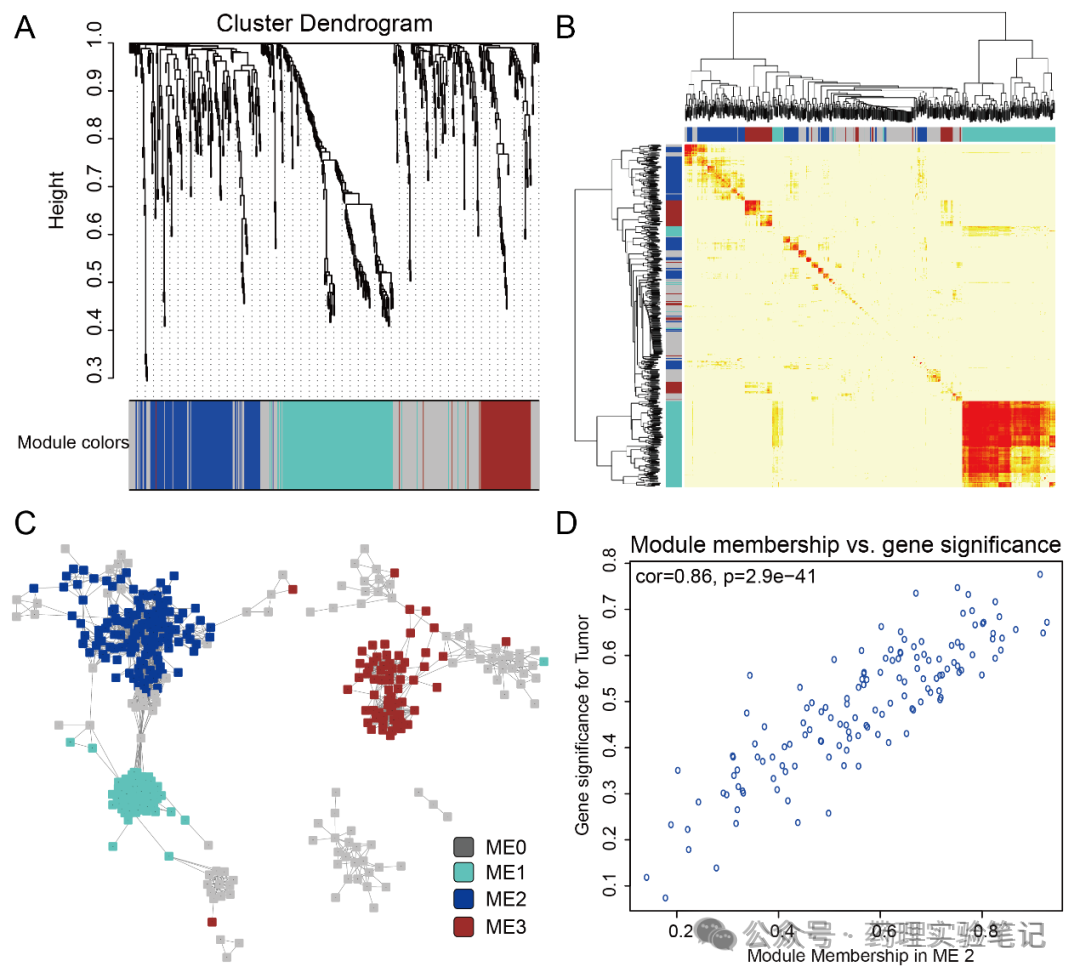
话不多说,直接上手。
一、准备数据
| 文件 | 格式 | 备注 |
|---|---|---|
| 表达矩阵 | 行为基因,列为样本 | csv |
| 分组信息 | 样本名分类, 0/1 | 1 是 treat,0 是 control |

library(WGCNA); library(tidyverse); library(limma)
exp <- read.csv("exp.csv", row.names = 1) # 表达矩阵
group <- read.table("group.txt", header = T, row.names = 1)
dat <- t(exp[tail(order(apply(exp,1,mad)),5000),]) # 取 MAD 前 5000原理:表达几乎不变(低 MAD)的基因,对"共表达"没贡献,还会把信号稀释掉。一刀切掉,既快又干净。只留下最活泼的 5000 个基因
二、去掉"捣乱"的样本 / 基因
gsg <- goodSamplesGenes(dat)
if(!gsg$allOK) dat <- dat[gsg$goodSamples, gsg$goodGenes]顺手画个树看看离群样本:
plot(hclust(dist(dat)), main = "Sample tree")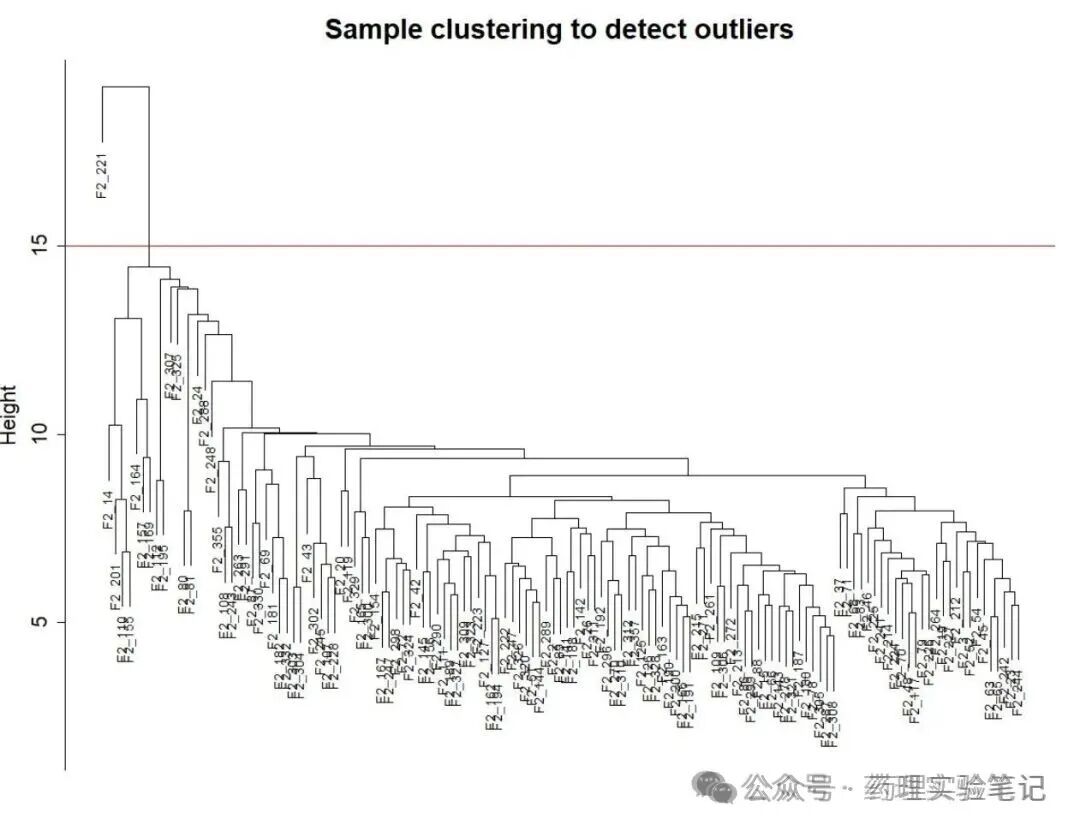
原理:坏样本------整体表达谱跟大队人马差太远,会把整棵树带歪。坏基因------空值太多或全零,拉低相关性矩阵质量。这一步相当于质检,省得后面白跑。
三、挑软阈值(power)
sft <- pickSoftThreshold(dat, powerVector = c(1:10, seq(12,30,2)))
power <- sft$powerEstimate # 直接拿来用
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2])
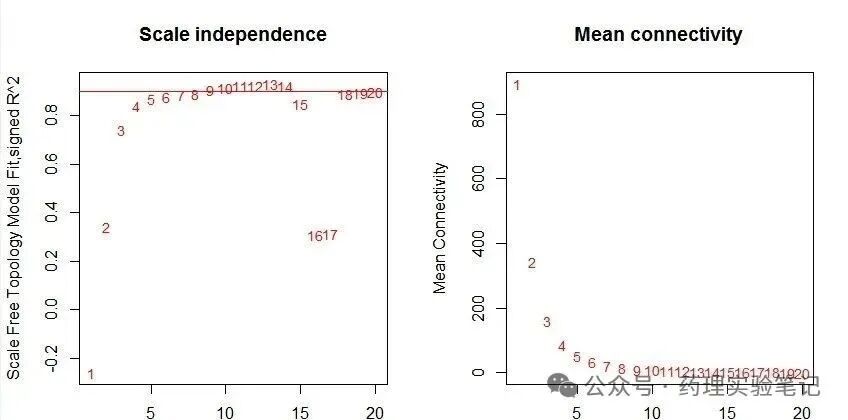
原理:原始相关系数是线性关系,经过幂次放大后,强相关更强,弱相关几乎归零。0.85 那条红线是"无标度"及格线,过了才承认"像生物网"。
四、建模块
-
power:上一步的 -
minModuleSize:模块最小基因数,默认 50,想多基因就调大 -
mergeCutHeight:模块合并阈值,默认 0.25,越小模块越多net <- blockwiseModules(
dat, power = power,
maxBlockSize = 5000,
minModuleSize = 75,
mergeCutHeight = 0.25,
numericLabels = TRUE)
看一眼模块大小:
table(net$colors)把模块画出来(2 张图)
moduleColors <- labels2colors(net$colors)
plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]])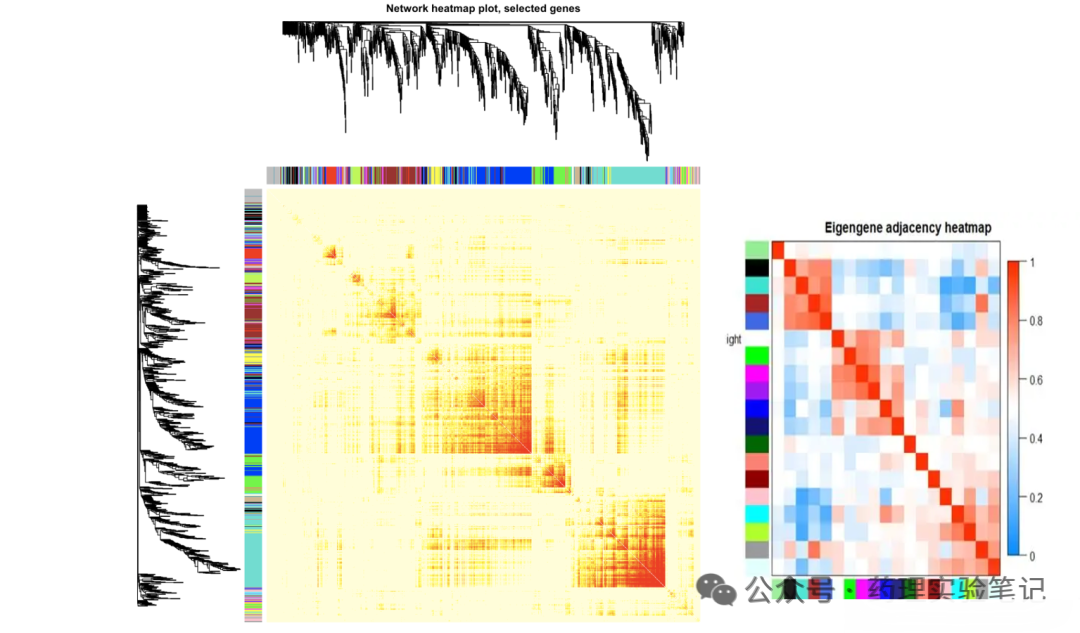
原理:minModuleSize / mergeCutHeight:调大 → 模块少且肥大;调小 → 模块多且瘦小。按需求拧螺丝即可。
五、模块 vs 表型
一句话:算模块特征基因(ME)跟分组 0/1 的相关性。
MEs <- net$MEs
colnames(MEs) <- paste0("ME", labels2colors(as.numeric(sub("ME","",colnames(MEs)))))
rownames(MEs) <- substr(rownames(MEs),1,12)
modTraitCor <- cor(MEs, group, use = "p")
modTraitP <- corPvalueStudent(modTraitCor, nrow(group))
labeledHeatmap(
Matrix = modTraitCor,
textMatrix = paste0(signif(modTraitCor,2),"\n(",signif(modTraitP,1),")"),
colors = colorRampPalette(c("blue","white","red"))(50))原理:每个模块用一条"代表基因"------ME(第一主成分)。ME 与 0/1 分组算相关,颜色越深、P 越小,说明整个模块"集体"跟疾病绑得越紧。
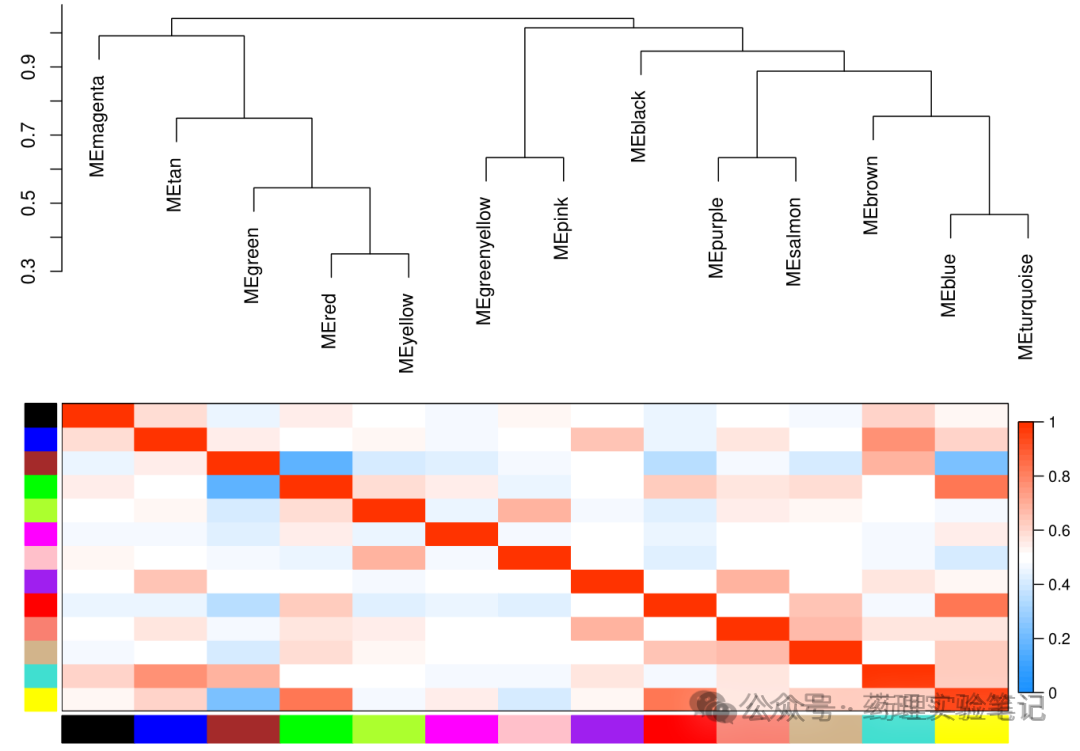
六、核心基因怎么筛?
假设最相关的是 blue 模块:
module <- "blue"
gene_exp <- dat[, net$blockGenes[[1]]]
ME <- MEs[, paste0("ME", module)]
# MM:基因与模块相关性
MM <- abs(cor(gene_exp, ME, use = "p"))
# GS:基因与表型相关性
GS <- abs(cor(gene_exp, group, use = "p"))
# 散点图看一眼
plot(MM, GS, pch = 19,
xlab = "MM (blue)", ylab = "Gene significance",
main = "MM vs GS")
# 取交集:MM & GS 双高
hub <- names(which(MM > 0.8 & GS > 0.5))hub 就是核心基因,拿去富集、验证、做实验.
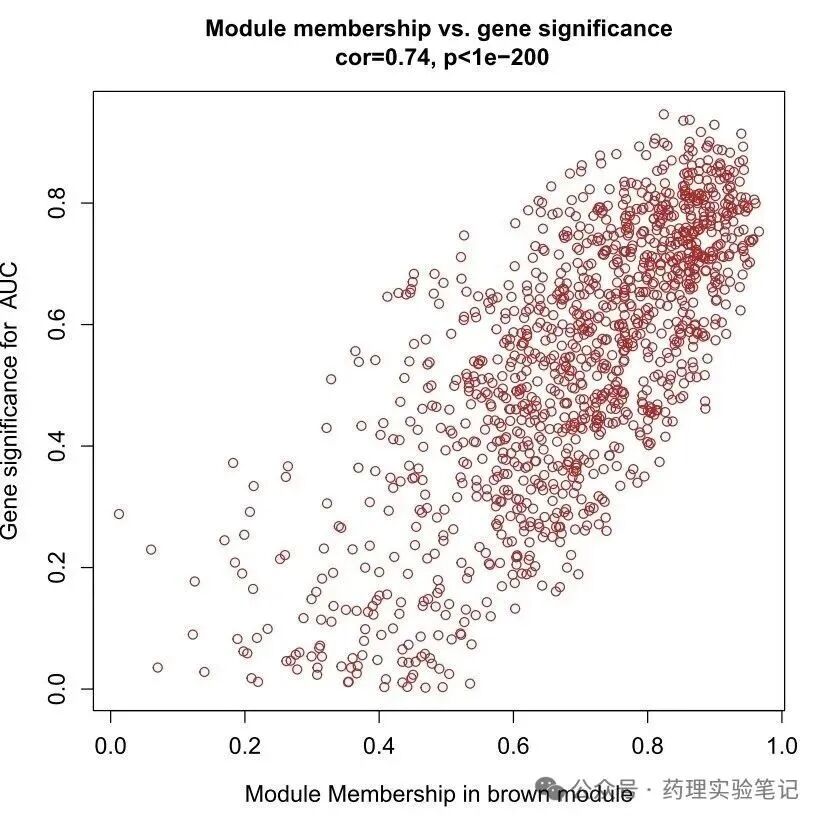
原理:MM:基因跟"群聊主题"的匹配度。GS:基因跟"疾病"本身的匹配度。两值都高 → 既是群核心,又跟疾病强相关。
七、导出 Cytoscape(可选)
TOM <- TOMsimilarityFromExpr(dat, power = power)
cyt <- exportNetworkToCytoscape(
TOM[hub, hub],
edgeFile = "edges.txt",
nodeFile = "nodes.txt",
weighted = TRUE, threshold = 0.2)
---------------------END---------------------
科研辅导与课题设计,欢迎联系我们!