Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning (Nature Machine Intelligence 2024 IS 23.9)
一、研究背景与模型架构
1.1 研究动机与背景
- RNA在基因表达、调控和催化等生物过程中起着关键作用。传统的RNA分析依赖于昂贵的实验技术(如RNA测序),而现有的计算方法(如RNA-FM, RNABERT)往往只针对特定任务,或者在预训练中仅使用简单的掩码策略,丢失了RNA序列中连续的高密度信息(如Motif)。此外,现有的预训练模型难以通过单一的权重集适应多种下游任务,且在面对训练中未见过的RNA序列分布时表现不佳。为了解决这些问题,📌作者提出了RNAErnie,旨在通过引入生物学先验知识来构建一个通用的RNA分析模型
1.2 模型架构与数据基础
- RNAErnie 基于ERNIE(Enhanced Representation through Knowledge Integration)框架构建,由12层Transformer组成,隐藏层维度为768。该模型在预训练阶段使用了来自RNAcentral数据库的约2300万条非编码RNA(ncRNA)序列
- 为了更好地处理RNA数据,模型将碱基(A, U, C, G)进行Token化,并在每个序列的末尾附加了粗粒度的RNA类型(如miRNA, lncRNA)作为特殊标记(Special Tokens)。这种设计不仅让模型学习序列信息,还能通过IND(指示符)标记学习将相似功能的RNA在潜在空间中聚类
二、核心创新方法
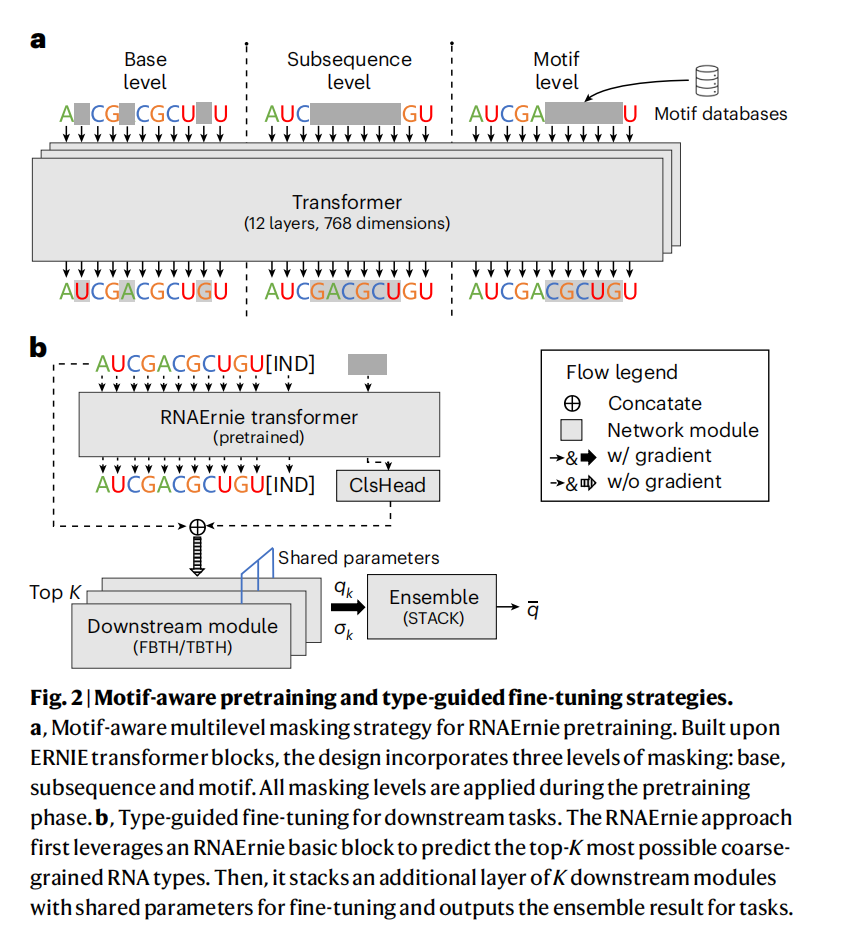
📌 2.1 Motif-Aware 的预训练策略
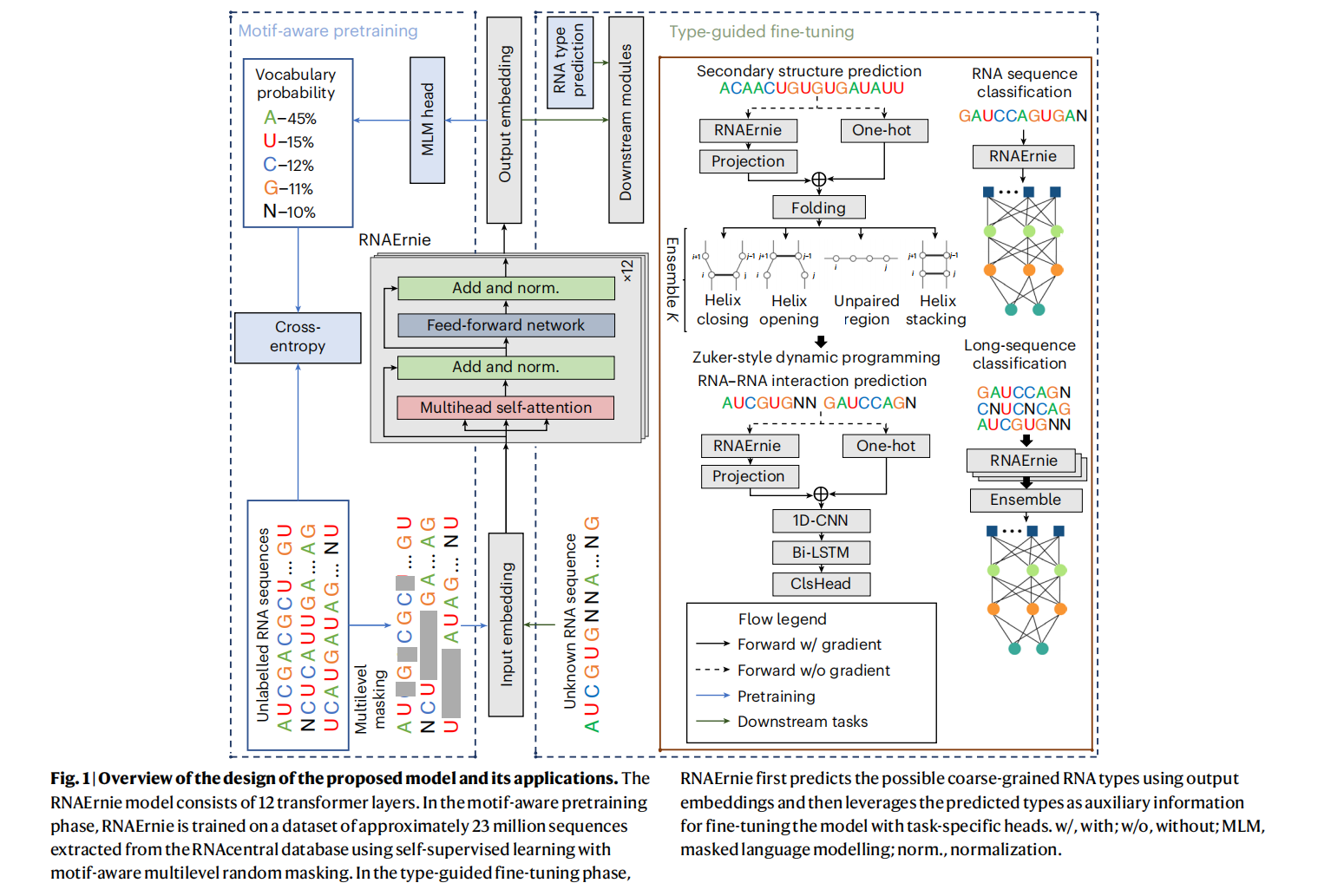
Step1: Multilevel Masking
RNAErnie 的核心创新之一是引入了多级掩码策略(Multilevel Masking),以捕获不同层级的生物学信息。这包括三个层次:
- 碱基级掩码(Base-level):随机掩盖15%的核苷酸,让模型学习基础的序列表示;
- 子序列级掩码(Subsequence-level):随机掩盖短的连续片段(4-8bp),迫使模型理解相邻碱基间的依赖关系;
- 模体级掩码(Motif-level):这是其独特之处,模型利用了来自ATTRACT、SpliceAid等数据库的生物学先验知识,针对具有特定生物功能的RNA模体(Motif)进行随机掩码。这使得模型能够捕捉到不仅限于一级序列结构的高级结构和功能信息
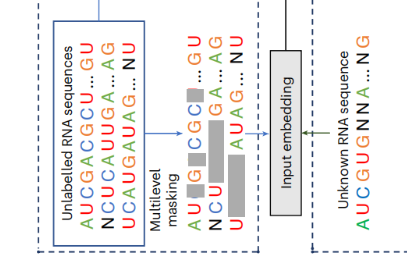
step 2: RNAErnie Transformer
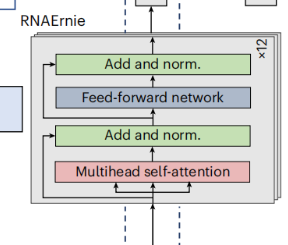
- 深入理解输入序列的上下文信息,将简单的碱基向量转化为蕴含丰富生物学意义的 Output embedding
step 3: 预测与概率分布
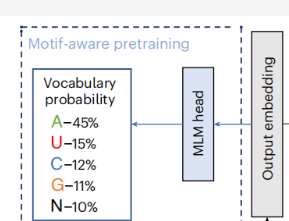
📌 MLM head(Masked Language Modeling Head):分类头,专门用来预测被掩码(Masked)位置的真实碱基
- Vocabulary probability:模型认为该位置是 A 的概率为 45%,是 U 的概率为 15%,以此类推。这表示模型根据上下文判断,这里最可能填入"A"
step 4: 误差计算与优化
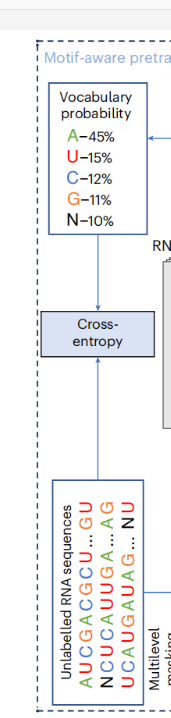
📌Cross-entropy(交叉熵损失函数):系统会将模型预测的概率(Vocabulary probability)与 原始的真实序列(Unlabelled RNA sequences) 进行对比
- 反向传播:这个误差信号会反向传回模型,调整 Transformer 和 Embedding 的参数,让模型在下一轮猜得更准
2.2 类型引导(Type-Guided)的微调策略
针对下游任务,特别是那些与预训练数据分布不同的任务,RNAErnie 提出了一种类型引导的微调策略(Stacking Architecture)。
-
该过程分为两步(Fig.2_b):首先,模型利用预训练的RNAErnie块预测输入序列可能属于的前K种粗粒度RNA类型(如mRNA, miRNA等)。其次,模型将这些预测出的类型作为辅助信息拼接到序列尾部,输入到K个并行的下游模块中进行微调,每个下游模块对应一个FBTH或TBTH,最后通过 Ensemble 得出最终结果。这种方法显著增强了模型在不同RNA类型和任务上的适应性
- FBTH(冻结主干),即冻结预训练的主干参数,仅训练任务特定的预测头。这种方法计算效率高,且在下游任务数据与预训练数据分布差异较大(域外分布)时表现更佳,能有效防止模型遗忘通用的生物学知识。
- TBTH(可训练主干),它对整个模型(主干和头)进行端到端的联合微调。当任务数据与预训练分布相似(域内分布)时,这种全参数更新策略能让模型深度适配特定任务特征,通常能获得最优的预测精度。
三、实验评估与应用
3.1 无监督学习表现
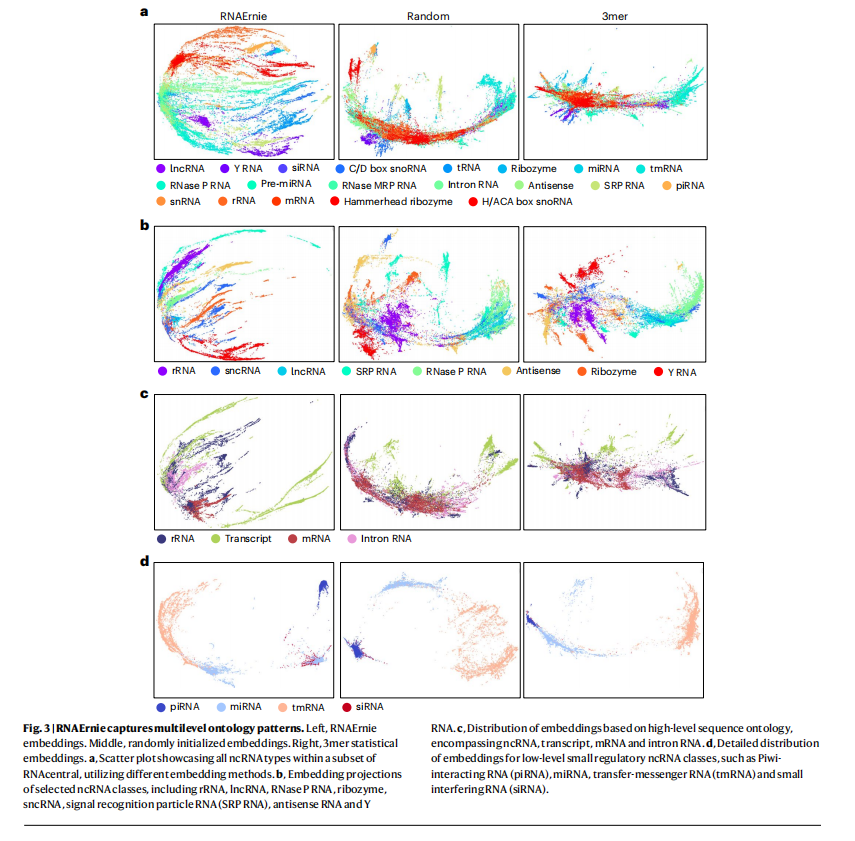
-
该研究利用无监督聚类方法来评估RNAErnie从原始序列中提取特征的有效性。为了直观地展示高维特征分布,研究采用了 PHATE 技术------这是一类专门用于保留生物数据全局结构和潜在轨迹的降维可视化算法,将复杂的嵌入向量映射到二维平面上。通过将RNAErnie生成的嵌入与随机初始化模型及传统的3-mer统计嵌入进行对比,研究者旨在验证该模型是否能自发捕捉到RNA的内在模式。
-
实验结果通过四个维度详细展示了模型的表征能力:
- 总体聚类(a)显示RNAErnie能基于结构和功能属性将所有已知RNA类型组织成界限分明的簇,相比之下,3-mer统计嵌入特征模糊不清,无法区分,而随机模型虽然有一定结构但定义较差
- 其次,在特定非编码RNA类别(b)上,模型生成的嵌入成功区分并聚集了rRNA、lncRNA、sncRNA以及核酶等主要类别,证明其捕获了超越一级序列的关键生物学特征
- 第三,在高层级本体(c)分析中,模型有效映射并分离了ncRNA、转录本、mRNA和内含子RNA及其相互关系,展示了对宏观序列功能的理解
- 最后,针对低层级小调节性RNA(d)(如miRNA、siRNA、piRNA),虽然模型能进行一定区分,但实验发现其在这一细粒度层面的辨识度不如随机初始化模型,这可能归因于此类RNA类别的高度异质性、复杂性或训练数据的潜在偏差,表明模型在捕捉微观调控模式上仍有优化空间
3.2 监督学习任务表现
RNAErnie 在多个监督学习基准任务上进行了广泛评估,均表现出色:
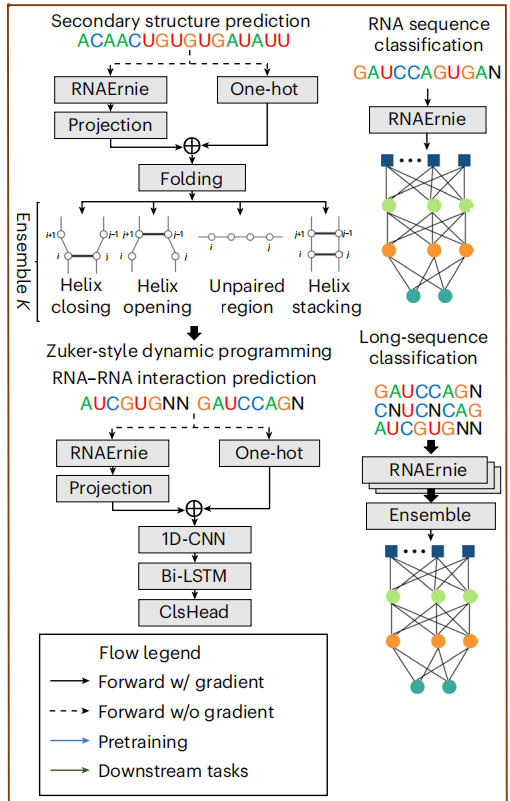
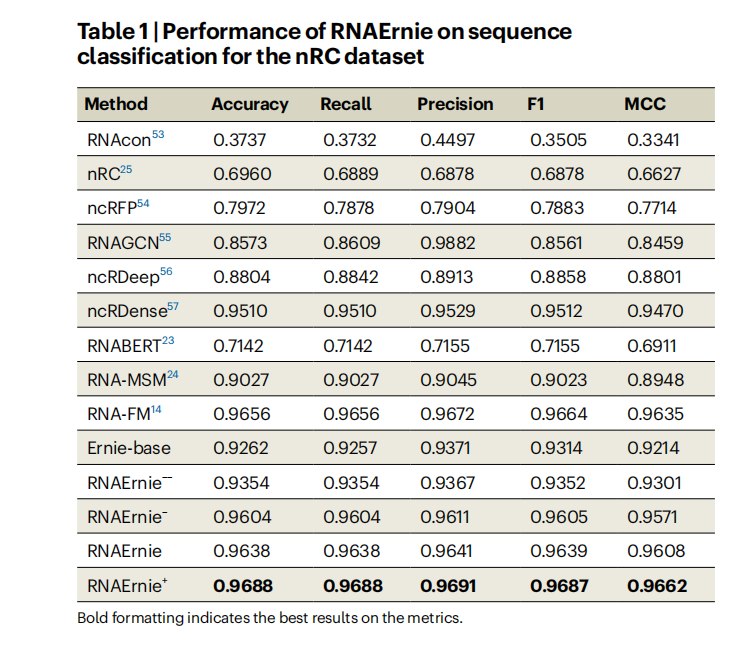
- 📌RNA序列分类:在nRC数据集上,RNAErnie及其变体在准确率、召回率和F1分数上均优于包括RNA-FM和RNABERT在内的基线模型
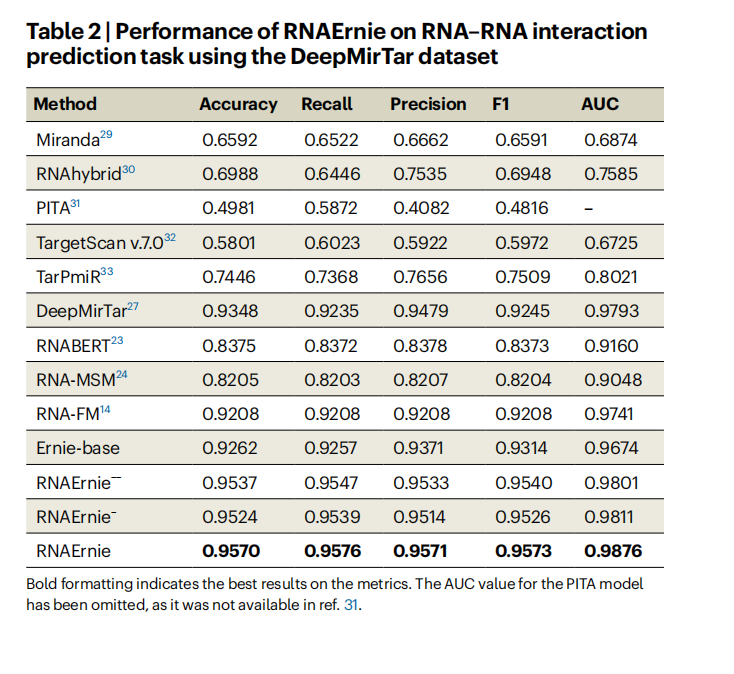
- 📌RNA-RNA相互作用预测:在DeepMirTar数据集上,RNAErnie展现了极高的预测能力,AUC达到0.9876,显著优于现有方法
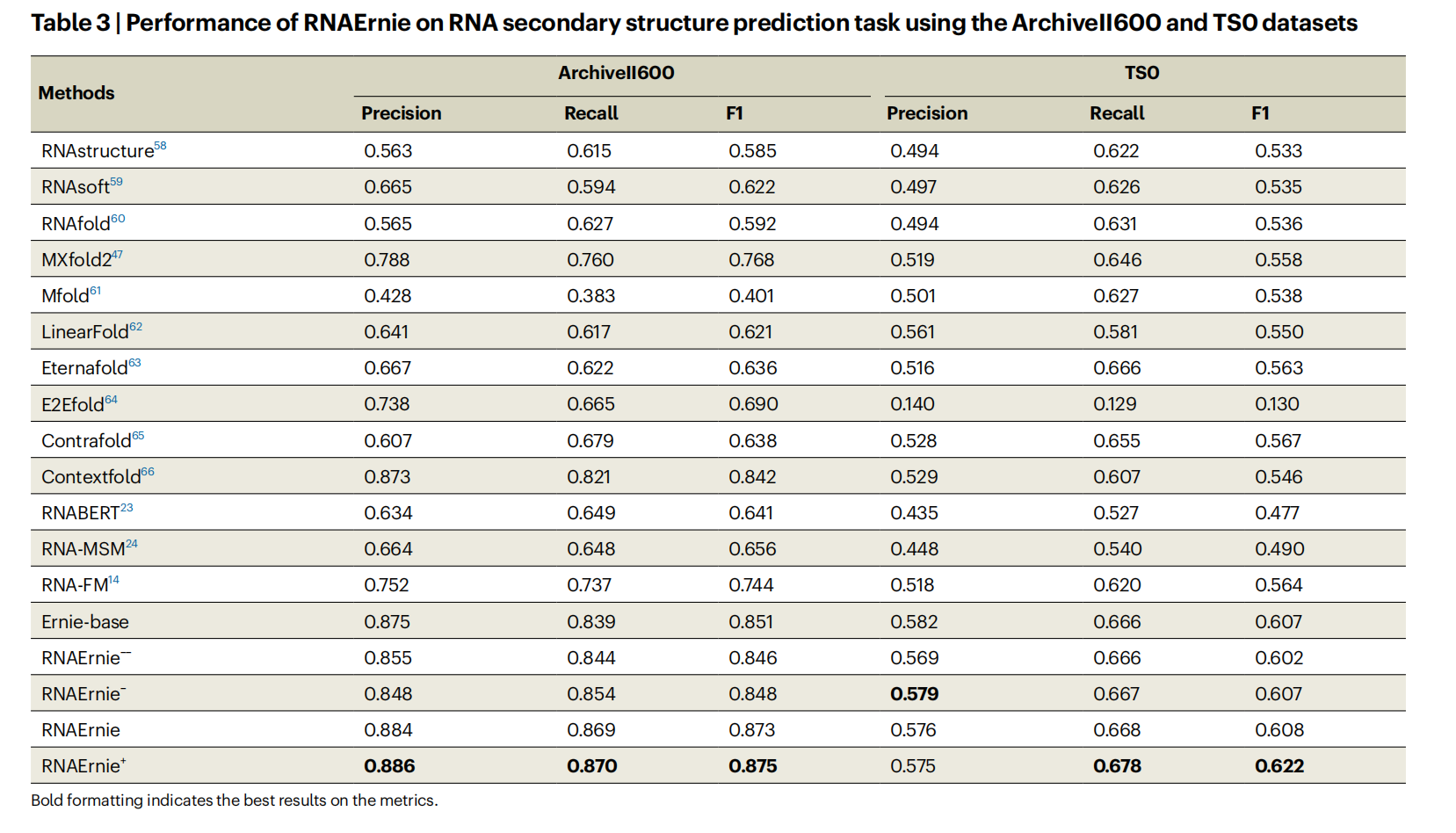
- 📌RNA二级结构预测:在ArchiveII和TSO数据集上,RNAErnie通过结合折叠算法,在精确度和F1分数上击败了UFold、LinearFold等专用工具
四、总结与局限性
4.1 总结
- RNAErnie 是一个多用途的RNA语言模型,它通过整合 Motif 感知的预训练和类型引导的微调策略,成功解决了单一模型适应多种RNA分析任务的难题。实验证明,它在分类、相互作用预测和结构预测等任务上均达到了 SOTA 的水平,展现了强大的鲁棒性和泛化能力
4.2 局限性与未来方向
尽管表现优异,RNAErnie 仍存在一些局限性:
- 序列长度限制:由于Transformer的计算复杂度,模型目前只能处理512个核苷酸以内的序列,长序列会被截断,导致部分长距离相互作用信息的丢失;
- 3D结构缺失:模型目前主要关注一维序列和二级结构,难以处理复杂的三维结构模体(如环和连接点);
- 推理开销:采用Stacking架构进行类型引导微调虽然提高了精度,但也增加了推理过程的计算开销。未来的工作可能会探索端到端的预训练方法来优化这一流程
⭐Github: https://github.com/CatIIIIIIII/RNAErnie
⭐文章地址:https://www.nature.com/articles/s42256-024-00836-4
⭐Modelscope: https://www.modelscope.cn/models/ZhejiangLab-LifeScience/rnaernie