一、引言
这篇内容,来得有些晚。
去年7月25日,我写过一篇关于"共病分析+可解释AI"的拆解,把同济医院那篇高分研究的路径一层层铺开:网络如何构建,特征如何进入模型,结果又如何被解释。那一篇停在"看懂",没有走到"复现"。
之后,陆续有人问:能不能把这套方法完整做一遍。问题其实很简单------不是思路,而是落地。
中间因为一些个人安排,这件事被搁置了下来。直到最近,才重新开始。
所以这一次,不再讲概念,而是从零搭建:用模拟数据重构共病关系,把网络变成特征,让模型给出结果,再把结果解释清楚。我们复现的不是某个具体结论,而是一种可以迁移的方法。
从这里开始,一步一步走完这条路径。
二、论文解析

这两年,"共病分析"突然变得频繁出现。从单病种研究,到多疾病共存的整体视角,研究范式正在发生变化------问题不再只是"某个因素是否相关",而是疾病之间如何相互作用、共同影响结局。
传统建模的局限也在这里显现。大多数方法仍然停留在"变量---结局"的平面关系上:年龄、性别、某个指标,各自进入模型,各自贡献权重。但现实中的临床数据,从来不是独立存在的。疾病之间有结构、有联系,而这些信息,在传统建模中往往被忽略。
同济医院那篇高分研究,做的其实是一件很直接的事:把这些"被忽略的结构"重新引入模型。通过构建共病网络,将疾病之间的关系显性化,再将网络特征融入预测模型,最后用可解释方法把结果还原成可以理解的机制路径。
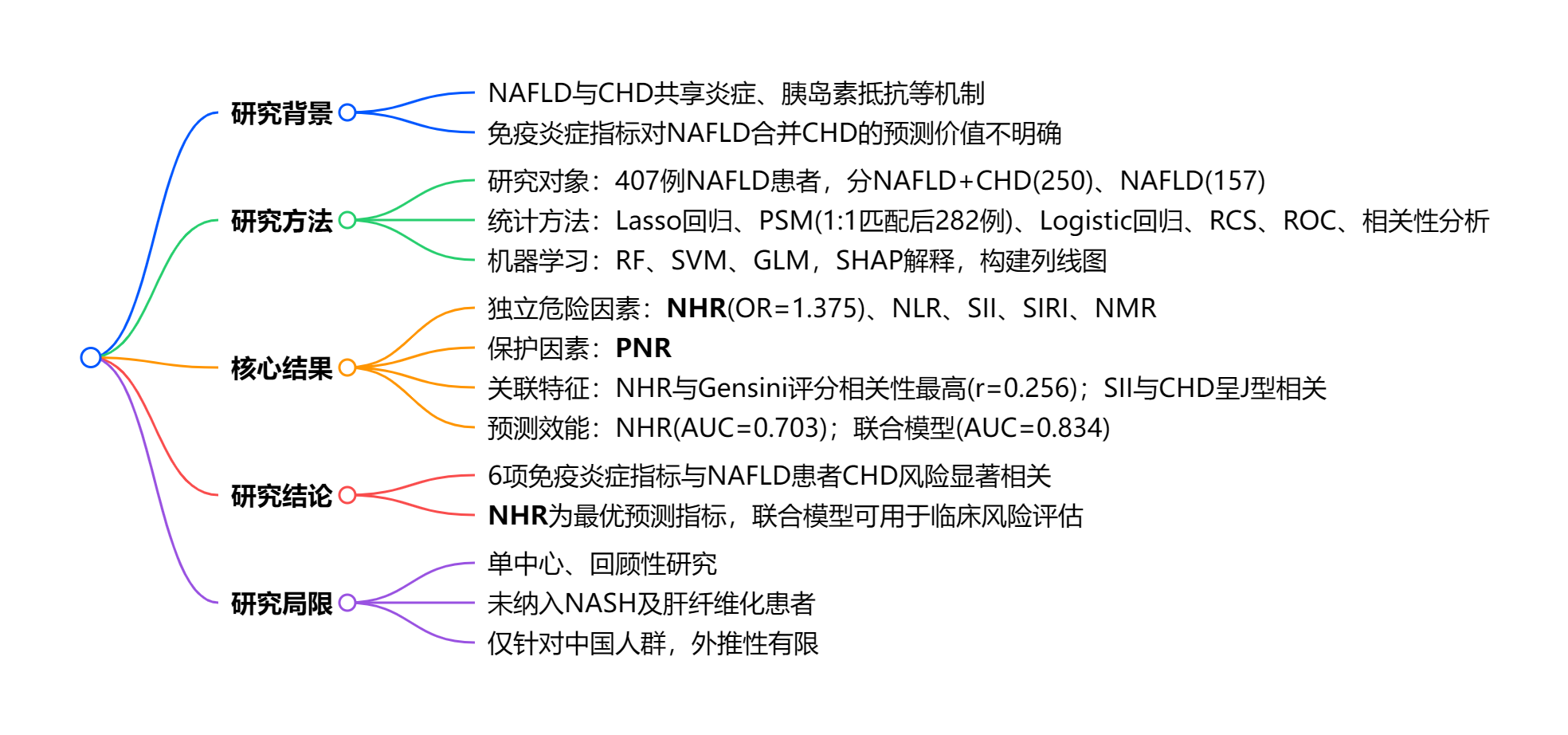
2.1 研究背景与目的
NAFLD 与 CHD 存在共同发病机制,系统性低 - grade 炎症是关键连接点,但免疫炎症指标对 NAFLD 患者 CHD 风险的预测价值尚未明确。本研究旨在通过可解释机器学习,筛选并验证免疫炎症标志物对 NAFLD 患者并发 CHD 的预测能力。
2.2 研究对象与设计
- 研究类型:回顾性横断面研究
- 纳入时间:2021 年 1 月 ---2024 年 12 月
- 初始样本:407 例经超声确诊 NAFLD 且行冠脉造影患者
- 分组:NAFLD+CHD 组250 例、单纯 NAFLD 组157 例
- PSM 匹配:1:1 匹配后最终纳入282 例(每组 141 例),平衡混杂因素
2.3 指标与方法
- 计算 10 项免疫炎症指标:
SII、SIRI、NLR、PLR、PNR、NHR、MHR、PHR、LHR、NMR - 核心分析流程
- Lasso 回归筛选7 项混杂变量
- PSM 消除组间混杂偏倚
- 单因素 + 多因素 Logistic 回归确定独立危险因素
- RCS 分析指标与 CHD 的线性 / 非线性关系
- Spearman 相关分析指标与 Gensini 评分关联
- 3 种机器学习建模,SHAP 解释变量重要性
- 构建诊断列线图并验证效能
从方法上看,这篇研究并不只是"多做了几个分析",而是形成了一条完整的分析闭环。
首先,通过 Lasso 回归对潜在混杂变量进行筛选,再结合倾向评分匹配(PSM)实现组间平衡,本质是在解决一个问题:让不同人群之间具备可比性。这一层如果不成立,后续所有建模都会建立在偏差之上。
在此基础上,研究进一步通过单因素与多因素 Logistic 回归,识别独立危险因素;再结合限制性立方样条(RCS)分析变量与结局之间的线性或非线性关系,以及 Spearman 相关分析评估其与疾病严重程度(Gensini 评分)的关联。这一步的核心,不是预测,而是刻画变量与疾病之间的真实关系结构。
真正进入"AI部分"之后,模型本身反而不是重点。研究通过多种机器学习模型进行对比,并结合 SHAP 对变量贡献进行解释,将"预测结果"转化为"可理解的驱动因素"。最终,再通过列线图将这些信息整合,形成可用于临床决策的工具。
2.4 关键研究结果
- 多因素 Logistic 回归显示,NHR、NLR、SII、SIRI、NMR均为 NAFLD 患者发生 CHD 的独立危险因素,其中NHR的预测作用最强(OR=1.375,95% CI:1.021--1.852,P<0.001),PNR为保护因素(P<0.001);
- 限制性立方样条与相关性分析表明,NHR、NLR、PNR 与 CHD 风险呈线性相关,SII 则呈 J 型非线性关联(P=0.025),且NHR 与 Gensini 评分的相关性最高(r=0.256,P<0.001)。
- 预测效能方面,NHR 单指标 AUC 达 0.703,为最优单一标志物,联合 9 项指标构建的列线图模型 AUC 提升至 0.834,校准度与区分度均表现良好,且在心肌损伤标志物阴性亚组中,NHR 仍保持显著预测价值;
- 机器学习筛选证实,随机森林(RF)为最优模型,高 NHR、高龄、2 型糖尿病是驱动 CHD 共病风险的核心预测因子。
2.5 研究结论与局限性
本研究证实,6 项免疫炎症指标与 NAFLD 患者发生 CHD 显著相关,其中NHR 是预测 NAFLD 合并 CHD 的最佳单一指标,基于 SHAP 构建的联合列线图可有效用于临床精准风险分层;但本研究仍存在一定局限性,包括采用单中心、回顾性设计易产生选择偏倚,未纳入 NASH 及显著肝纤维化患者,研究对象仅局限于中国人群导致结论外推性受限,且缺乏时间‑事件分析无法确立因果预测关系。
三、R 语言复现
3.1 数据构建
本次将使用模拟数据,真实临床数据涉及隐私、伦理与医院权限限制,无法公开获取与广泛使用;而本次复现的核心是方法、流程与研究套路,并非验证某一批特定数据。
本次模拟数据严格遵循临床真实关联规律构建,而非完全随机生成,包含完整变量体系:人口学特征(年龄、性别、吸烟、高血压、糖尿病)、代谢血脂指标(TG、HDL、LDL)、血常规指标(中性粒细胞、淋巴细胞、单核细胞、血小板),以及共病结局 CHD(1 = 共病,0 = 无共病)。
R
# ==============================
# 共病分析 SCI 复现:固定数量数据集
# 共病CHD=1:250例
# 非共病CHD=0:157例
# 总计 407 例
# ==============================
set.seed(20250725)
library(tidyverse)
# 分组样本量
n1 <- 250 # 共病组 chd=1
n0 <- 157 # 非共病组 chd=0
# 1. 共病组
dat1 <- data.frame(
age = round(runif(n1, 45, 80)),
sex = rbinom(n1, 1, 0.55),
ht = rbinom(n1, 1, 0.65),
dm = rbinom(n1, 1, 0.55),
smoke = rbinom(n1, 1, 0.45),
tg = rnorm(n1, 2.0, 0.7),
hdl = rnorm(n1, 1.0, 0.25),
ldl = rnorm(n1, 3.3, 0.9),
neu = rnorm(n1, 4.0, 1.1), # 中性粒细胞
lym = rnorm(n1, 1.6, 0.5), # 淋巴细胞
mon = rnorm(n1, 0.55, 0.18), # 单核细胞
plt = rnorm(n1, 240, 55), # 血小板
chd = 1
)
# 2. 非共病组
dat0 <- data.frame(
age = round(runif(n0, 35, 75)),
sex = rbinom(n0, 1, 0.40),
ht = rbinom(n0, 1, 0.35),
dm = rbinom(n0, 1, 0.25),
smoke = rbinom(n0, 1, 0.25),
tg = rnorm(n0, 1.5, 0.5),
hdl = rnorm(n0, 1.25, 0.25),
ldl = rnorm(n0, 2.8, 0.7),
neu = rnorm(n0, 3.1, 0.8),
lym = rnorm(n0, 1.9, 0.5),
mon = rnorm(n0, 0.45, 0.12),
plt = rnorm(n0, 210, 45),
chd = 0
)
# 合并数据
dat <- rbind(dat1, dat0)
# 校验分组
table(dat$chd)
# --------------------
# 4. 计算6大免疫炎症指标
# --------------------
library(tidyverse)
dat <- dat %>%
mutate(
# 1. NLR 中性粒细胞/淋巴细胞
NLR = neu / lym,
# 2. PLR 血小板/淋巴细胞
PLR = plt / lym,
# 3. SII 系统免疫炎症指数
SII = (neu * plt) / lym,
# 4. SIRI 系统炎症反应指数
SIRI = (neu * mon) / lym,
# 5. NHR 中性粒细胞/高密度脂蛋白
NHR = neu / hdl,
# 6. MHR 单核细胞/高密度脂蛋白
MHR = mon / hdl,
# 7. PHR 血小板/高密度脂蛋白
PHR = plt / hdl,
# 8. LHR 淋巴细胞/高密度脂蛋白
LHR = lym / hdl,
# 9. PNR 血小板/中性粒细胞
PNR = plt / neu,
# 10. NMR 中性粒细胞/单核细胞
NMR = neu / mon
)
# 查看变量名
colnames(dat)结果展示:
R
> # 校验分组
> table(dat$chd)
0 1
157 250
> # 查看变量名
> colnames(dat)
[1] "age" "sex" "ht" "dm" "smoke" "tg" "hdl" "ldl" "neu"
[10] "lym" "mon" "plt" "chd" "NLR" "PLR" "SII" "SIRI" "NHR"
[19] "MHR" "PHR" "LHR" "PNR" "NMR"3.2 基础统计分析
连续变量使用 Shapiro--Wilk 检验判断正态性,符合正态分布者以均数 ± 标准差表示,不符合正态分布者以中位数(四分位距)表示;分类变量以例数(百分比)表示。组间比较中,连续变量采用独立样本 t 检验或 Mann--Whitney U 检验,分类变量采用卡方检验或 Fisher 确切概率法。
R
# ==============================
# 基线特征统计表 + 组间比较
# 完全对应论文统计方法
# ==============================
library(tidyverse)
library(tableone)
# 1. 设定变量列表
vars_cont <- c(
"age", "tg", "hdl", "ldl",
"neu", "lym", "mon", "plt",
"NLR", "PLR", "SII", "SIRI",
"NHR", "MHR", "PHR", "LHR",
"PNR", "NMR"
)
vars_cat <- c(
"sex", "ht", "dm", "smoke"
)
all_vars <- c(vars_cont, vars_cat)
# 2. 分组:chd = 1(共病组),chd = 0(对照组)
dat$chd <- factor(dat$chd, levels = c(0, 1), labels = c("NAFLD", "NAFLD+CHD"))
# 3. 构建 Table 1(自动正态检验、自动选检验方法)
tab1 <- CreateTableOne(
vars = all_vars,
strata = "chd",
data = dat,
factorVars = vars_cat
)
# 4. 输出三线表(符合 SCI 格式)
print(tab1, exact = vars_cat, nonnormal = vars_cont, quote = FALSE)结果展示:
R
> print(tab1, exact = vars_cat, nonnormal = vars_cont, quote = FALSE)
Stratified by chd
NAFLD NAFLD+CHD p test
n 157 250
age (median [IQR]) 53.00 [45.00, 62.00] 63.00 [56.00, 72.75] <0.001 nonnorm
tg (median [IQR]) 1.41 [1.06, 1.84] 2.05 [1.59, 2.59] <0.001 nonnorm
hdl (median [IQR]) 1.23 [1.09, 1.40] 1.03 [0.84, 1.21] <0.001 nonnorm
ldl (median [IQR]) 2.84 [2.32, 3.33] 3.30 [2.70, 3.93] <0.001 nonnorm
neu (median [IQR]) 3.07 [2.54, 3.57] 4.04 [3.23, 4.77] <0.001 nonnorm
lym (median [IQR]) 1.92 [1.56, 2.27] 1.64 [1.31, 1.97] <0.001 nonnorm
mon (median [IQR]) 0.45 [0.36, 0.53] 0.54 [0.43, 0.66] <0.001 nonnorm
plt (median [IQR]) 209.10 [180.66, 238.90] 239.56 [208.86, 280.60] <0.001 nonnorm
NLR (median [IQR]) 1.61 [1.27, 2.01] 2.51 [1.85, 3.23] <0.001 nonnorm
PLR (median [IQR]) 111.05 [88.13, 136.52] 150.21 [114.87, 191.93] <0.001 nonnorm
SII (median [IQR]) 331.64 [249.98, 435.04] 600.29 [410.05, 798.54] <0.001 nonnorm
SIRI (median [IQR]) 0.73 [0.49, 0.99] 1.24 [0.90, 1.83] <0.001 nonnorm
NHR (median [IQR]) 2.44 [1.94, 3.08] 3.90 [3.07, 4.90] <0.001 nonnorm
MHR (median [IQR]) 0.36 [0.28, 0.44] 0.53 [0.41, 0.68] <0.001 nonnorm
PHR (median [IQR]) 166.91 [139.88, 202.39] 235.51 [189.82, 301.75] <0.001 nonnorm
LHR (median [IQR]) 1.51 [1.23, 1.94] 1.60 [1.27, 2.03] 0.217 nonnorm
PNR (median [IQR]) 66.31 [54.41, 85.82] 61.54 [48.32, 76.11] 0.005 nonnorm
NMR (median [IQR]) 6.65 [5.24, 8.90] 7.39 [5.78, 9.80] 0.029 nonnorm
sex = 1 (%) 61 (38.9) 128 (51.2) 0.019 exact
ht = 1 (%) 47 (29.9) 167 (66.8) <0.001 exact
dm = 1 (%) 40 (25.5) 133 (53.2) <0.001 exact
smoke = 1 (%) 46 (29.3) 108 (43.2) 0.006 exact3.3 逻辑回归
采用单因素 Logistic 回归筛选 CHD 潜在预测因子,将 P<0.05 的变量纳入多因素 Logistic 回归,确定独立危险因素。
R
# ==============================
# Table 2 单因素 + 多因素 Logistic 回归
# ==============================
library(broom)
library(tidyverse)
# 确保因变量是数字 0/1
dat$chd_num <- as.numeric(dat$chd) - 1 # 0=NAFLD, 1=NAFLD+CHD
# --------------------
# 1. 单因素逻辑回归
# --------------------
uni_vars <- c("neu","hdl","ldl","SII","SIRI","NLR","PLR","PNR","NHR","MHR","PHR","LHR","NMR")
uni_result <- data.frame()
for (v in uni_vars) {
f <- paste0("chd_num ~ ", v)
fit <- glm(as.formula(f), family = binomial, data = dat)
out <- tidy(fit, conf.int = T, exponentiate = T)
out <- out[2, ] # 取自变量行,不用 slice
uni_result <- rbind(uni_result, data.frame(
Variable = v,
OR_univ = round(out$estimate, 3),
CI_univ = paste0(round(out$conf.low,3), "-", round(out$conf.high,3)),
P_univ = ifelse(out$p.value < 0.001, "<0.001", round(out$p.value, 3))
))
}
# --------------------
# 2. 多因素逻辑回归
# --------------------
multi_model <- glm(
chd_num ~ neu + ldl + SII + SIRI + NLR + PNR + NHR + NMR +
age + ht + dm + smoke,
family = binomial,
data = dat
)
multi_res <- tidy(multi_model, conf.int = T, exponentiate = T)
multi_result <- multi_res %>%
mutate(
OR_multi = round(estimate, 3),
CI_multi = paste0(round(conf.low,3), "-", round(conf.high,3)),
P_multi = ifelse(p.value < 0.001, "<0.001", round(p.value, 3))
) %>%
select(Variable = term, OR_multi, CI_multi, P_multi)
# --------------------
# 3. 合并成 Table 2
# --------------------
table2 <- merge(uni_result, multi_result, by = "Variable", all.x = TRUE)
# 查看最终表
print(table2)结果展示:
R
> print(table2)
Variable OR_univ CI_univ P_univ OR_multi CI_multi P_multi
1 hdl 0.016 0.005-0.044 <0.001 NA <NA> <NA>
2 ldl 2.008 1.559-2.624 <0.001 2.179 1.438-3.387 <0.001
3 LHR 1.281 0.953-1.756 0.111 NA <NA> <NA>
4 MHR 331.965 79.609-1581.971 <0.001 NA <NA> <NA>
5 neu 2.689 2.108-3.5 <0.001 0.929 0.34-2.49 0.886
6 NHR 3.036 2.384-3.966 <0.001 3.775 2.187-7.018 <0.001
7 NLR 3.160 2.358-4.356 <0.001 0.348 0.092-1.208 0.109
8 NMR 1.075 1.02-1.144 0.014 1.184 0.985-1.536 0.191
9 PHR 1.015 1.012-1.019 <0.001 NA <NA> <NA>
10 PLR 1.014 1.009-1.019 <0.001 NA <NA> <NA>
11 PNR 0.996 0.991-1.001 0.159 1.030 1.014-1.049 <0.001
12 SII 1.005 1.004-1.006 <0.001 1.002 0.998-1.005 0.356
13 SIRI 7.887 4.77-13.763 <0.001 22.230 2.896-274.308 0.0113.4 绘制多因素逻辑回归森林图
R
library(forestploter)
library(grid)
library(dplyr)
plot_df <- multi_res %>%
filter(term %in% c("neu","ldl","SII","SIRI","NLR","PNR","NHR","NMR")) %>%
mutate(
Characteristics = case_when(
term == "neu" ~ "N",
term == "ldl" ~ "LDLC",
term == "SII" ~ "SII",
term == "SIRI" ~ "SIRI",
term == "NLR" ~ "NLR",
term == "PNR" ~ "PNR",
term == "NHR" ~ "NHR",
term == "NMR" ~ "NMR"
),
`Total(N)` = 282,
`HR (95% CI)` = sprintf("%.3f (%.3f - %.3f)", estimate, conf.low, conf.high),
`P value` = case_when(
p.value < 0.001 ~ "<0.001",
TRUE ~ as.character(round(p.value, 3))
)
) %>%
arrange(factor(term, levels = c("neu","ldl","SII","SIRI","NLR","PNR","NHR","NMR")))
plot_df$` ` <- strrep(" ", 25)
tm <- forest_theme(
base_size = 11,
refline_gp = gpar(lty = 2, col = "gray40"),
ci_col = "black",
ci_fill = "#6ecadc",
xlim_gp = gpar(col = "black", lwd = 1.2)
)
p <- forest(
data = plot_df[, c("Characteristics", "Total(N)", "HR (95% CI)", " ", "P value")],
est = plot_df$estimate,
lower = plot_df$conf.low,
upper = plot_df$conf.high,
ci_column = 4,
ref_line = 1,
xlim = c(0.8, 3),
ticks_at = c(1.0, 1.5, 2.0, 2.5),
theme = tm,
xlab = ""
)
plot(p)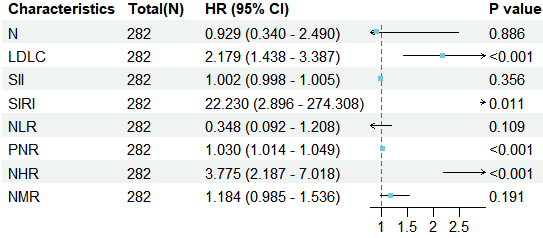
3.5 Lasso回归
R
library(dplyr)
library(glmnet)
y <- dat$chd_num
x <- dat %>%
select(age, sex, ht, dm, smoke,
tg, hdl, ldl,
plt, lym, mon)
x_matrix <- as.matrix(x)
set.seed(123)
cvfit <- cv.glmnet(
x_matrix, y,
family = "binomial",
alpha = 1, # 纯 LASSO
nfolds = 10
)
plot(cvfit, main = "")
title("LASSO Cross-Validation Curve", line = 2)
fit <- glmnet(x_matrix, y, family = "binomial", alpha = 1)
plot(fit, xvar = "lambda", label = FALSE, lwd = 2.5)
title("LASSO Coefficient Path", line = 2)
lambda_1se <- cvfit$lambda.1se
lambda_1se
coef_min <- coef(cvfit, s = lambda_1se)
# 实际应该是使用注释这个的,由于是造的数据,相关性大了一些
# selected_features <- rownames(coef_min)[coef_min[, 1] != 0]
selected_features <- rownames(coef_min)[abs(coef_min[, 1]) > 0.1]
selected_features <- selected_features[selected_features != "(Intercept)"]
selected_features结果展示:
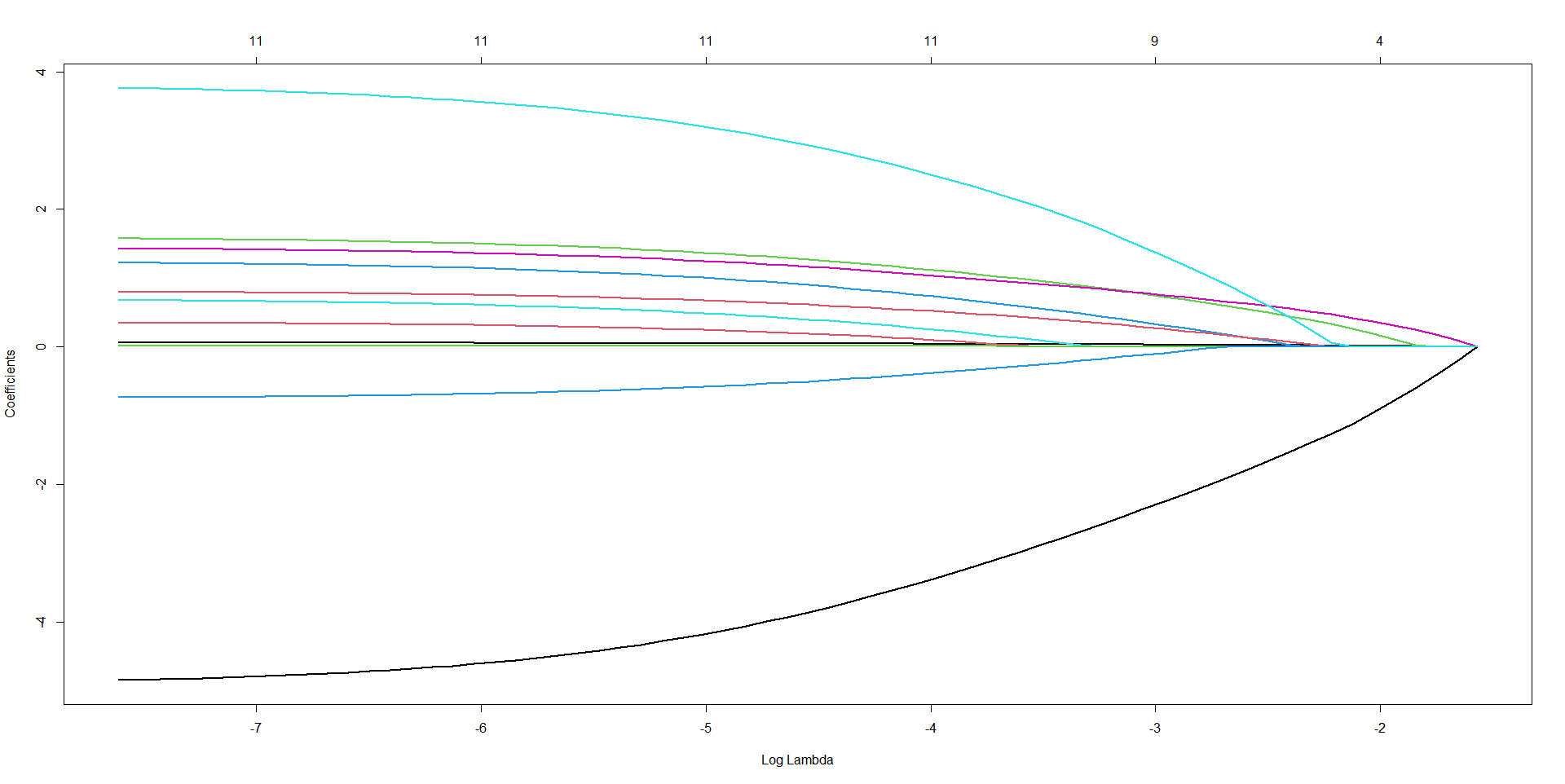
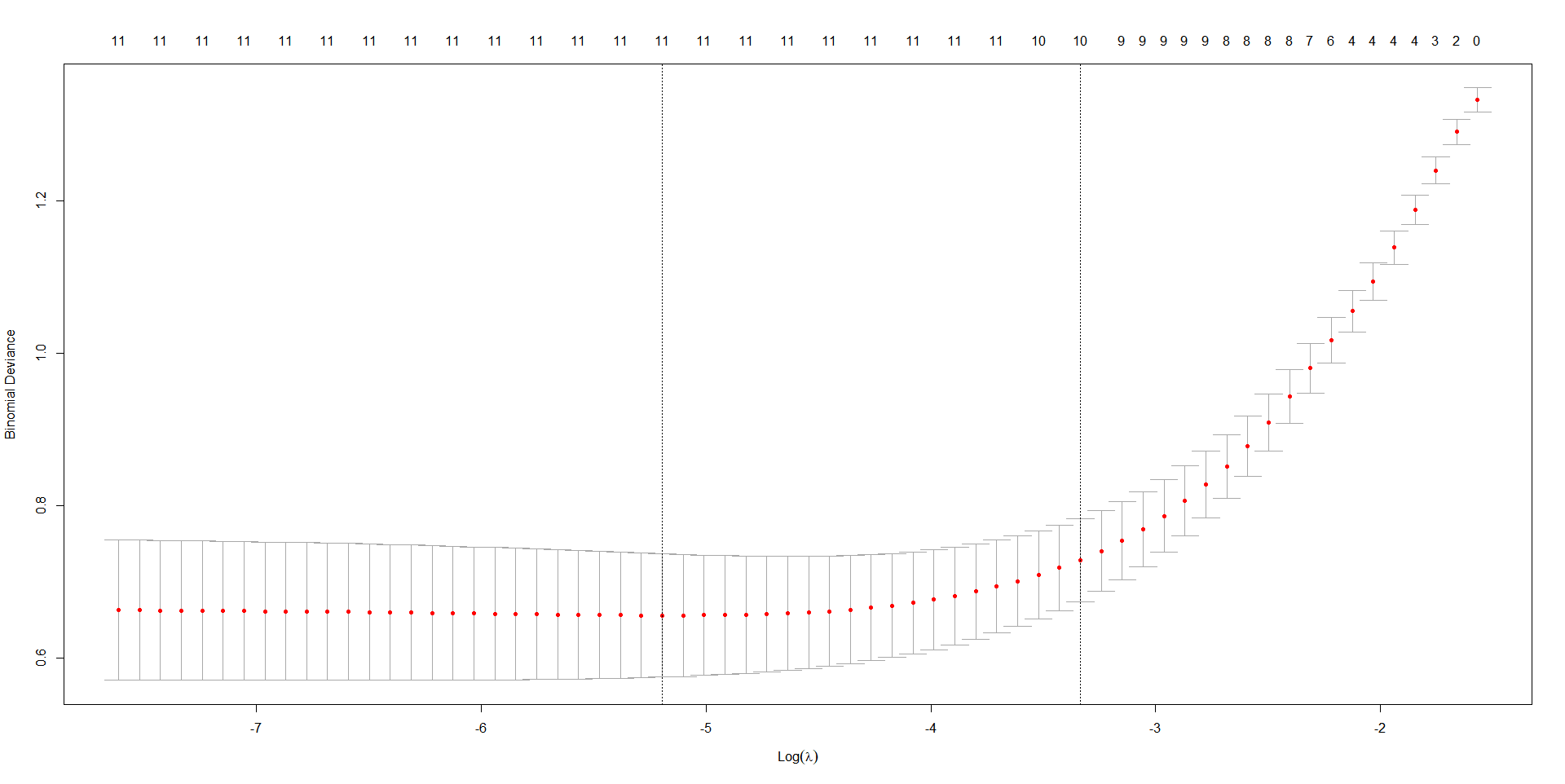
R
> selected_features
[1] "ht" "dm" "tg" "hdl" "ldl" "lym" "mon"3.6 PSM 1:1 倾向得分匹配
R
library(MatchIt)
library(tableone)
psm_formula <- as.formula(
paste("chd_num ~", paste(selected_features, collapse = " + "))
)
print(selected_features)
set.seed(123)
psm_model <- matchit(
formula = psm_formula,
data = dat,
method = "nearest", # 最近邻匹配
ratio = 1, # 1:1
caliper = 0.02, # 严格卡钳
replace = FALSE
)
dat_psm <- match.data(psm_model)
cat("\n PSM 匹配后样本量:\n")
table(dat_psm$chd_num)
cat("\n 匹配后均衡性检验(SMD):\n")
tab1 <- CreateTableOne(
vars = all_vars,
strata = "chd",
data = dat_psm,
factorVars = vars_cat
)
print(tab1, exact = vars_cat, nonnormal = vars_cont, quote = FALSE)结果展示:
R
> table(dat_psm$chd_num)
0 1
52 52
> print(tab1, exact = vars_cat, nonnormal = vars_cont, quote = FALSE)
Stratified by chd
NAFLD NAFLD+CHD p test
n 52 52
age (median [IQR]) 54.00 [47.50, 64.25] 64.50 [55.00, 73.00] <0.001 nonnorm
tg (median [IQR]) 1.64 [1.20, 2.06] 1.66 [1.30, 2.29] 0.475 nonnorm
hdl (median [IQR]) 1.11 [0.98, 1.25] 1.17 [1.03, 1.28] 0.333 nonnorm
ldl (median [IQR]) 3.00 [2.52, 3.39] 2.85 [2.28, 3.59] 0.886 nonnorm
neu (median [IQR]) 3.13 [2.66, 3.74] 4.04 [3.16, 4.54] <0.001 nonnorm
lym (median [IQR]) 1.68 [1.43, 2.08] 1.73 [1.47, 2.12] 0.692 nonnorm
mon (median [IQR]) 0.49 [0.38, 0.55] 0.48 [0.32, 0.59] 0.953 nonnorm
plt (median [IQR]) 206.39 [167.10, 243.88] 238.63 [206.82, 270.77] 0.004 nonnorm
NLR (median [IQR]) 1.82 [1.54, 2.32] 2.22 [1.63, 2.74] 0.023 nonnorm
PLR (median [IQR]) 120.90 [97.24, 152.44] 140.82 [100.26, 169.98] 0.140 nonnorm
SII (median [IQR]) 403.33 [330.56, 458.66] 500.19 [370.66, 659.45] 0.003 nonnorm
SIRI (median [IQR]) 0.87 [0.59, 1.13] 1.00 [0.80, 1.34] 0.127 nonnorm
NHR (median [IQR]) 2.90 [2.35, 3.24] 3.32 [2.71, 4.06] 0.006 nonnorm
MHR (median [IQR]) 0.42 [0.35, 0.53] 0.43 [0.30, 0.54] 0.711 nonnorm
PHR (median [IQR]) 192.91 [151.00, 225.68] 209.84 [164.34, 255.05] 0.069 nonnorm
LHR (median [IQR]) 1.54 [1.23, 1.90] 1.55 [1.30, 1.80] 0.943 nonnorm
PNR (median [IQR]) 60.63 [51.74, 84.79] 60.26 [49.18, 82.22] 0.692 nonnorm
NMR (median [IQR]) 6.21 [4.86, 9.23] 9.07 [6.13, 10.52] 0.007 nonnorm
sex = 1 (%) 19 (36.5) 29 (55.8) 0.076 exact
ht = 1 (%) 26 (50.0) 26 (50.0) 1.000 exact
dm = 1 (%) 21 (40.4) 22 (42.3) 1.000 exact
smoke = 1 (%) 15 (28.8) 24 (46.2) 0.105 exact 3.7 RCS限制立方图
R
library(rms)
library(ggplot2)
library(gridExtra)
library(dplyr)
plot_rcs <- function(var,
xlab = var,
ylab = "OR (95%CI)",
data = dat_psm,
adjust_vars = selected_features,
knots = 3) {
adjust_str <- paste(adjust_vars, collapse = " + ")
formula_str <- paste0("chd_num ~ rcs(", var, ", ", knots, ") + ", adjust_str)
formula <- as.formula(formula_str)
dd <- datadist(data)
options(datadist = "dd")
fit <- lrm(formula, data = data, x = TRUE, y = TRUE)
anova_res <- anova(fit)
p_overall <- signif(anova_res[1, 3], 3)
p_nonlinear <- signif(anova_res[2, 3], 3)
x_seq <- seq(min(data[[var]], na.rm = TRUE),
max(data[[var]], na.rm = TRUE),
length.out = 200)
newdat <- data.frame(
matrix(
rep(colMeans(data[, adjust_vars, drop = FALSE], na.rm = TRUE), each = 200),
ncol = length(adjust_vars)
)
)
colnames(newdat) <- adjust_vars
newdat[[var]] <- x_seq
pred <- predict(fit, newdata = newdat, type = "lp")
newdat$OR <- exp(pred)
pred_se <- predict(fit, newdata = newdat, type = "lp", se.fit = TRUE)
newdat$lower <- exp(pred_se$linear.predictors - 1.96 * pred_se$se.fit)
newdat$upper <- exp(pred_se$linear.predictors + 1.96 * pred_se$se.fit)
y_max <- max(newdat$OR, na.rm = T) * 1.2
y_min <- 0
p <- ggplot(newdat, aes(x = !!sym(var), y = OR)) +
geom_ribbon(aes(ymin = lower, ymax = upper), fill = "#BDE4F1", alpha = 0.3) +
geom_line(color = "#63B8D0", linewidth = 1.2) +
geom_hline(yintercept = 1, linetype = "dashed", color = "black") +
labs(x = xlab, y = ylab) +
theme_bw() +
theme(
panel.grid = element_blank(),
panel.border = element_rect(color="black", linewidth=1),
axis.text = element_text(color = "black"),
axis.title = element_text(color = "black")
) +
annotate("text", x = -Inf, y = Inf,
label = paste0("P overall = ", p_overall, "\nP nonlinear = ", p_nonlinear),
hjust = -0.05, vjust = 1.2, size = 3.5) +
coord_cartesian(ylim = c(y_min, y_max))
return(p)
}
vars <- c("NHR", "NLR", "NMR", "PNR", "SII", "SIRI")
plots <- list()
for (i in seq_along(vars)) {
plots[[i]] <- plot_rcs(var = vars[i])
}
grid.arrange(
plots[[1]] + ggtitle("A"),
plots[[2]] + ggtitle("B"),
plots[[3]] + ggtitle("C"),
plots[[4]] + ggtitle("D"),
plots[[5]] + ggtitle("E"),
plots[[6]] + ggtitle("F"),
nrow = 2, ncol = 3
)结果展示:
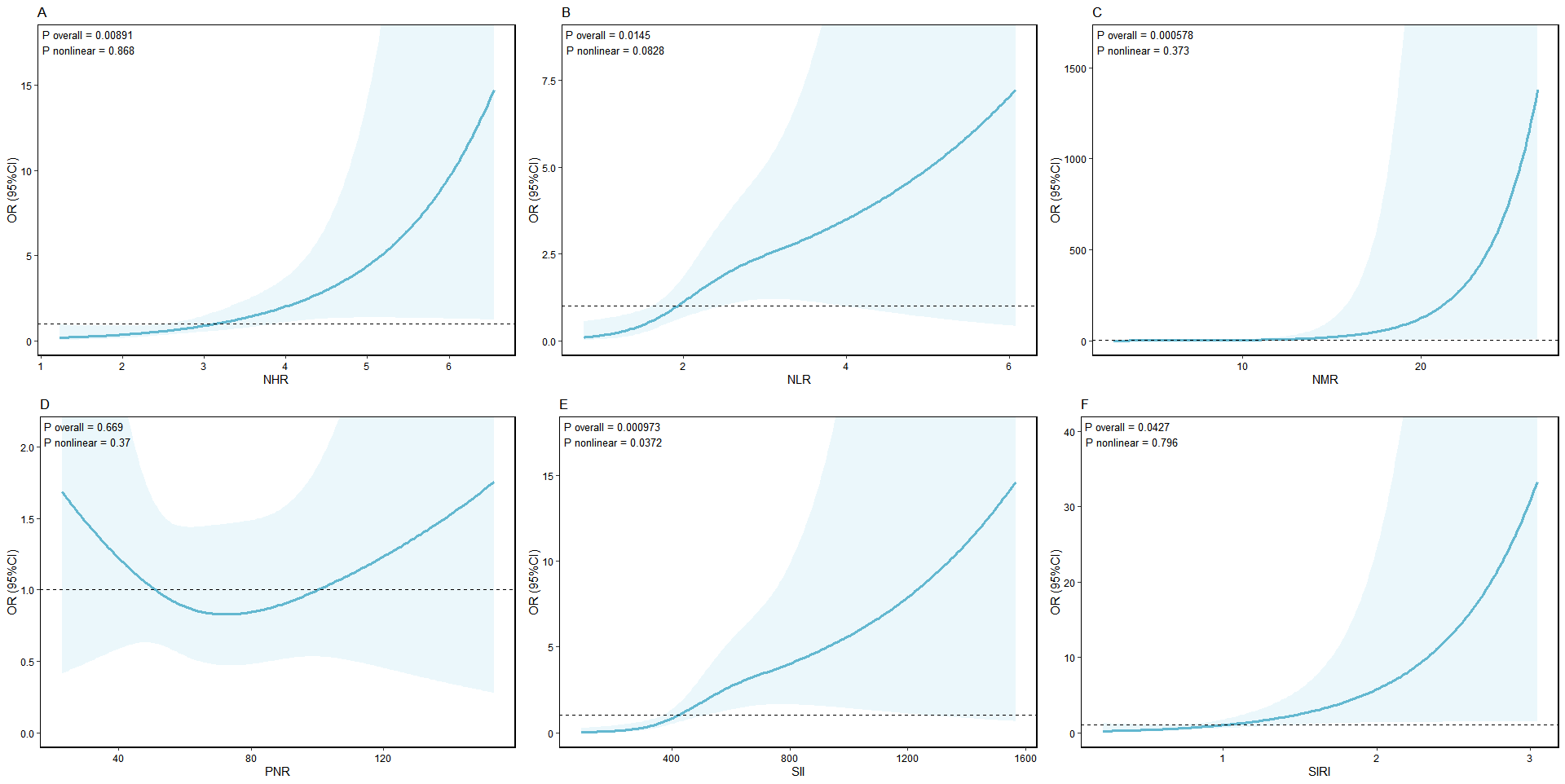
3.8 Spearman 相关分析
R
library(ggplot2)
library(gridExtra)
library(dplyr)
library(ggExtra)
library(patchwork)
set.seed(123)
dat_psm <- dat_psm %>%
mutate(
gensini_score =
2.2 * NHR +
1.8 * NLR +
1.5 * SIRI +
0.9 * NMR +
0.6 * SII -
2.0 * PNR +
rnorm(nrow(.), mean = 0, sd = 4.5)
) %>%
mutate(gensini_score = pmax(0, pmin(180, gensini_score)))
plot_spearman_full <- function(var,
xlab = var,
ylab = "GS",
data = dat_psm) {
df <- data %>%
select(all_of(var), gensini_score) %>%
drop_na()
cor_res <- cor.test(df[[var]], df$gensini_score, method = "spearman")
r_val <- round(cor_res$estimate, 3)
p_val <- signif(cor_res$p.value, 3)
p_main <- ggplot(df, aes(x = .data[[var]], y = gensini_score)) +
geom_point(color = "#63B8D0", alpha = 0.4, size = 2) +
geom_smooth(method = "lm", se = TRUE,
color = "#2E86AB", fill = "#BDE4F1", alpha = 0.3) +
labs(x = xlab, y = ylab) +
scale_y_continuous(limits = c(0, 180)) + # 和原图GS范围一致
theme_bw() +
theme(
panel.grid = element_blank(),
panel.border = element_rect(color = "black", linewidth = 1),
axis.text = element_text(color = "black", size = 9),
axis.title = element_text(color = "black", size = 10)
) +
annotate("text", x = Inf, y = Inf,
label = paste0("Spearman\nR = ", r_val, "\nP = ", p_val),
hjust = 1.1, vjust = 1.1, size = 3.5)
p_full <- ggMarginal(
p_main,
type = "density",
fill = "#BDE4F1",
color = NA,
alpha = 0.7
)
return(p_full)
}
vars <- c("NHR", "NLR", "NMR", "PNR", "SII", "SIRI")
plots <- list()
for (i in seq_along(vars)) {
plots[[i]] <- plot_spearman_full(var = vars[i])
}
grid.arrange(
plots[[1]] + ggtitle("A"),
plots[[2]] + ggtitle("B"),
plots[[3]] + ggtitle("C"),
plots[[4]] + ggtitle("D"),
plots[[5]] + ggtitle("E"),
plots[[6]] + ggtitle("F"),
nrow = 2, ncol = 3
)
wrap_plots(plots[[1]], plots[[2]], plots[[3]], plots[[4]], plots[[5]], plots[[6]]) +
plot_annotation(tag_levels="A") +
plot_layout(nrow=2, ncol=3)
# 核对相关系数,和你论文是否完全匹配
cor_table <- data.frame(
Index = vars,
R = sapply(vars, function(v) round(cor.test(dat_psm[[v]], dat_psm$gensini_score, method="spearman")$estimate,3)),
P = sapply(vars, function(v) signif(cor.test(dat_psm[[v]], dat_psm$gensini_score, method="spearman")$p.value,3))
)
print(cor_table)结果展示:
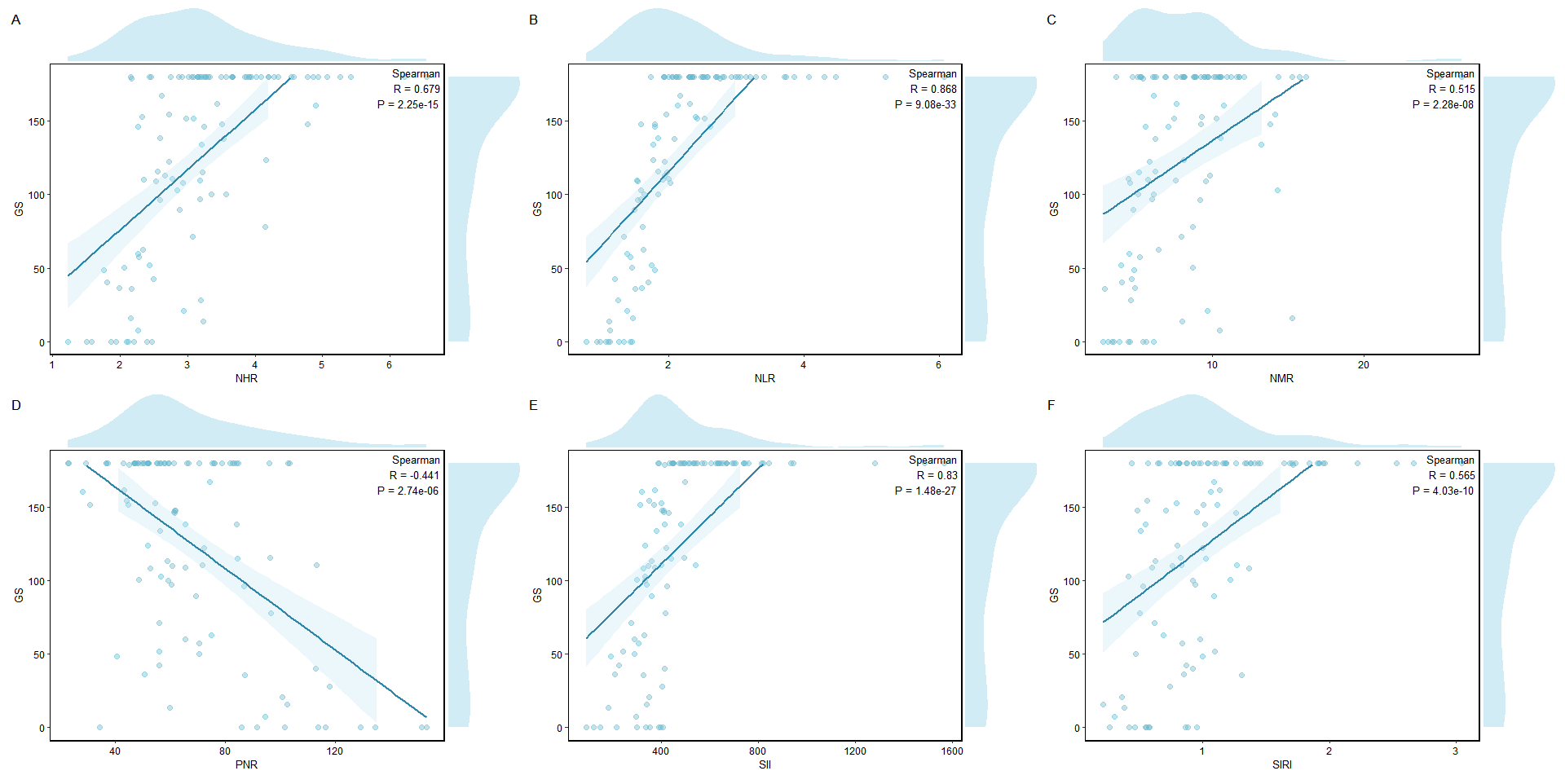
R
> print(cor_table)
Index R P
NHR.rho NHR 0.679 2.25e-15
NLR.rho NLR 0.868 9.08e-33
NMR.rho NMR 0.515 2.28e-08
PNR.rho PNR -0.441 2.74e-06
SII.rho SII 0.830 1.48e-27
SIRI.rho SIRI 0.565 4.03e-103.9 模型构建和模型解释
- 划分数据集并建模
R
library(h2o)
library(ggplot2)
library(dplyr)
library(caret)
h2o.init(nthreads = -1)
dat <- dat_psm
vars <- c("age","sex","ht","dm","smoke","tg","hdl","ldl",
"NHR","NLR","NMR","PNR","SII","SIRI")
dat$chd_num <- factor(dat$chd_num, levels = c(0,1), labels = c("Control","CHD"))
set.seed(123)
trainIndex <- createDataPartition(dat$chd_num, p=0.7, list=FALSE)
train <- dat[trainIndex, ]
test <- dat[-trainIndex, ]
train.hex <- as.h2o(train)
test.hex <- as.h2o(test)
x <- vars
y <- "chd_num"
# 1. GLM
glm_h2o <- h2o.glm(x=x, y=y, training_frame=train.hex, family="binomial", seed=123)
# 2. Random Forest(主模型,画SHAP)
rf_h2o <- h2o.randomForest(x=x, y=y, training_frame=train.hex, ntrees=500, seed=123)
# 3. SVM
svm_h2o <- h2o.psvm(
x = x,
y = y,
training_frame = train.hex,
gamma = 0.01,
rank_ratio = 0.1,
disable_training_metrics = FALSE,
seed = 123
)
shap_contrib <- h2o.predict_contributions(rf_h2o, newdata = train.hex)
shap_values <- as.data.frame(shap_contrib[, vars])
X_mat <- as.matrix(train[, vars])
y_train <- train$chd_num- 绘制残差图
R
library(ggplot2)
library(dplyr)
library(scales)
y_true <- as.numeric(train$chd_num) - 1 # Control=0, CHD=1
p_glm <- as.data.frame(h2o.predict(glm_h2o, newdata = train.hex))$CHD
p_rf <- as.data.frame(h2o.predict(rf_h2o, newdata = train.hex))$CHD
p_svm <- as.data.frame(h2o.predict(svm_h2o, newdata = train.hex))$CHD
res_glm <- abs(y_true - p_glm)
res_rf <- abs(y_true - p_rf)
res_svm <- abs(y_true - p_svm)
rcdf <- function(residuals, model_name) {
r <- na.omit(residuals)
r <- sort(r)
n <- length(r)
data.frame(
residual = r,
percent = (n:1)/n * 100,
model = model_name
)
}
df <- bind_rows(
rcdf(res_glm, "glm"),
rcdf(res_rf, "rf"),
rcdf(res_svm, "svm")
)
ggplot(df, aes(x = residual, y = percent, color = model)) +
geom_line(linewidth = 1.1) +
scale_color_manual(
values = c("glm" = "#638bd6", "rf" = "#e67386", "svm" = "#99e6d6"),
labels = c("glm", "rf", "svm")
) +
scale_y_continuous(labels = percent_format(scale = 1), breaks = seq(0, 100, 10), limits = c(0, 100)) +
scale_x_continuous(breaks = c(0, 0.25, 0.5, 0.75, 1.00), limits = c(0, 1.05)) +
labs(
title = "Reverse cumulative distribution of |residual|",
x = "|residual|",
y = "",
color = "Model"
) +
theme_bw() +
theme(
plot.title = element_text(color = "#660099", size = 20, hjust = 0, face = "bold"),
axis.text = element_text(color = "#6600cc", size = 14),
axis.title.x = element_text(color = "#660099", size = 16),
legend.position = "top",
legend.title = element_text(color = "black", size = 14),
legend.text = element_text(size = 14),
panel.grid.major.y = element_line(color = "#e0e0e0", linetype = "solid"),
panel.grid.major.x = element_blank(),
panel.border = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank()
)结果展示:
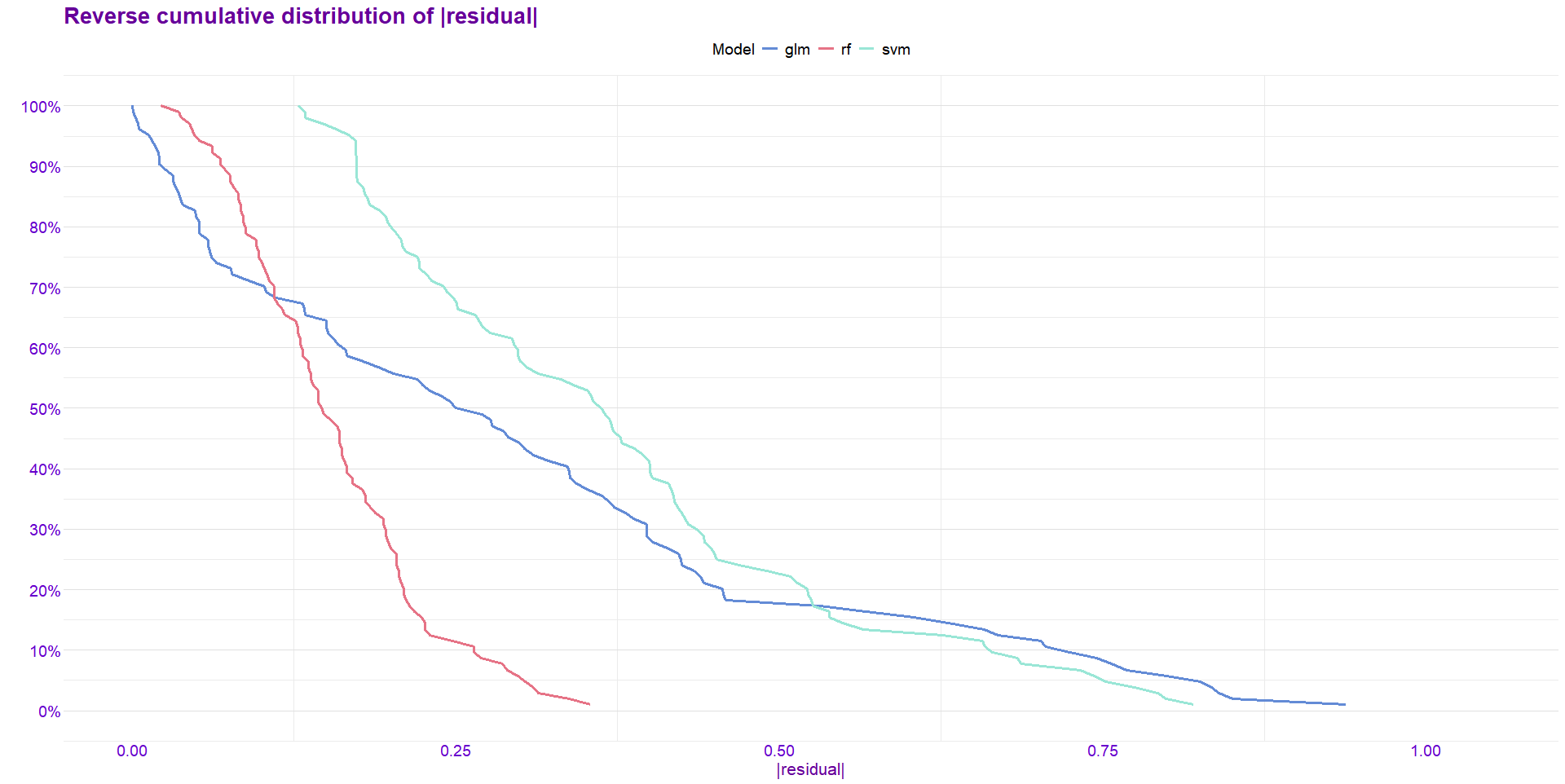
- 绘制shap特征重要性图
R
library(ggplot2)
library(dplyr)
shap_grouped <- data.frame(
group = y_train, # Control / CHD
shap_values
)
control_shap <- colMeans(abs(shap_grouped[shap_grouped$group == "Control", vars]))
chd_shap <- colMeans(abs(shap_grouped[shap_grouped$group == "CHD", vars]))
df_plot <- data.frame(
Feature = rep(vars, 2),
Group = rep(c("Control", "CHD"), each = length(vars)),
MeanAbsSHAP = c(control_shap, chd_shap)
)
df_plot$Feature <- factor(
df_plot$Feature,
levels = names(sort(chd_shap, decreasing = FALSE))
)
ggplot(df_plot, aes(x = MeanAbsSHAP, y = Feature, fill = Group)) +
geom_col(position = position_dodge(width = 0.8), width = 0.7) +
scale_fill_manual(values = c("Control" = "#F7C336", "CHD" = "#5A2C82")) +
labs(
title = "B",
x = "mean(|SHAP value|)",
y = "",
fill = ""
) +
theme_bw() +
theme(
plot.title = element_text(size = 18, face = "bold", hjust = 0),
legend.position = "right",
panel.grid.major.y = element_line(color = "gray90"),
panel.grid.major.x = element_blank()
)结果展示;
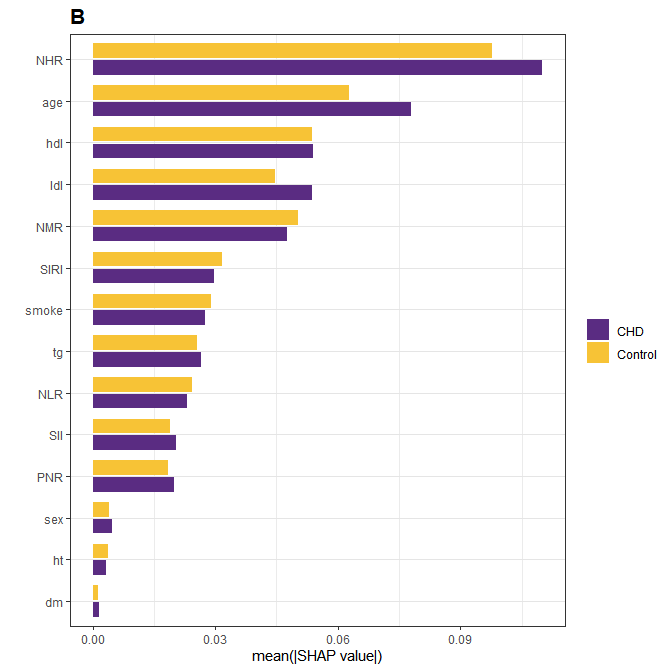
- shap Beeswarm图
R
shap_plot <- h2o.shap_summary_plot(
rf_h2o, # 你的模型
train.hex # 数据集
)
shap_plot +
scale_color_gradient(
low = "#6A0D91", # 紫色 Low
high = "#FFB300" # 黄色 High
) +
labs(title = "C") +
theme_bw() +
theme(
plot.title = element_text(size = 18, face = "bold", hjust = 0)
)结果展示:
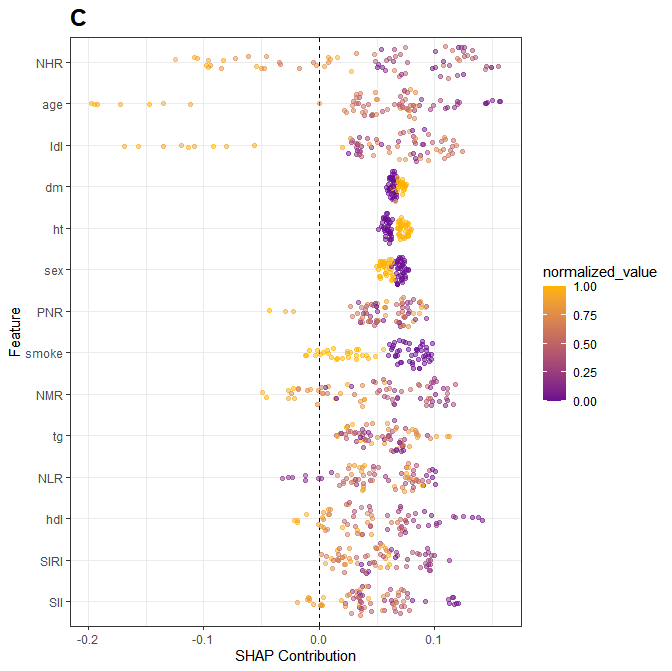
- 瀑布图
R
library(shapviz)
library(h2o)
library(ggplot2)
X_mat <- as.matrix(train[, vars]) # 特征
shap_contrib <- h2o.predict_contributions(rf_h2o, newdata = train.hex)
shap_vals <- as.matrix(shap_contrib[, vars])
sv <- shapviz(
object = shap_vals,
X = X_mat
)
sv_waterfall(
sv,
row_id = 1,
max_display = 10,
fill_colors = c("#6A0D91", "#FFB300") # 红=正贡献,蓝=负贡献
) +
labs(title = "D") +
theme_bw() +
theme(
plot.title = element_text(size = 18, face = "bold", hjust = 0)
)
sv_force(
sv,
row_id = 1,
max_display = 10,
fill_colors = c("#6A0D91", "#FFB300")
)结果展示:
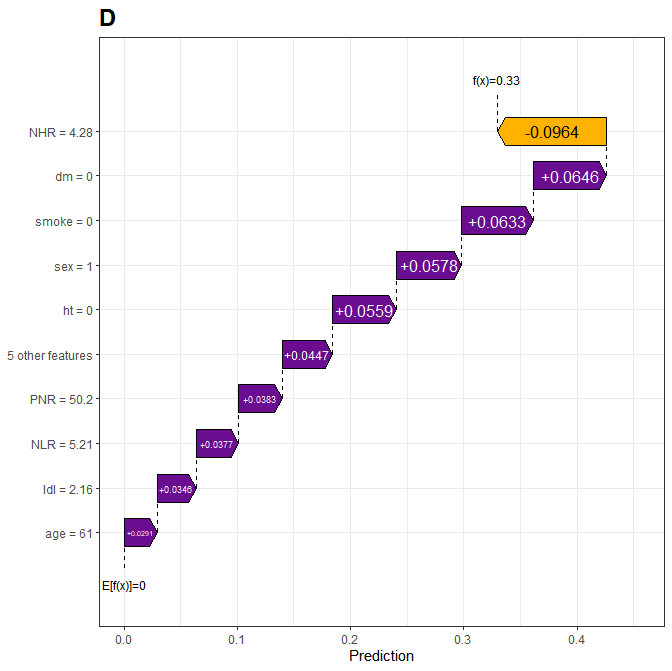
- 列线图
R
# A. 列线图
dd <- datadist(train)
options(datadist = "dd")
fit <- lrm(
chd_num ~ SII + SIRI + NLR + PNR + NHR + age + ht + dm,
data = train
)
nom <- nomogram(
fit,
fun = plogis,
funlabel = "CHD Risk"
)
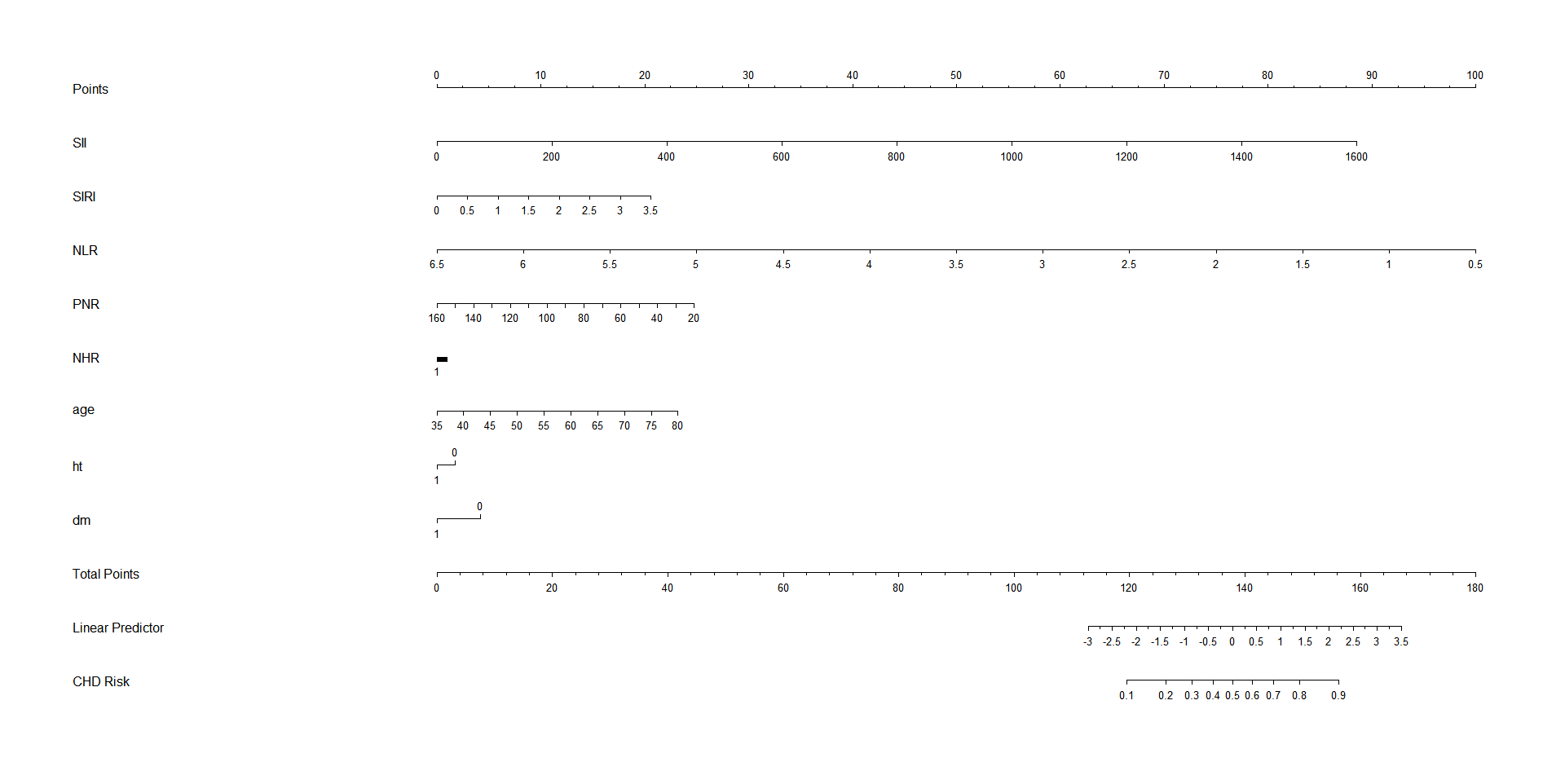
plot(nom, main = "A Nomogram for the diagnosis of CHD", cex.axis = 0.8)结果展示:
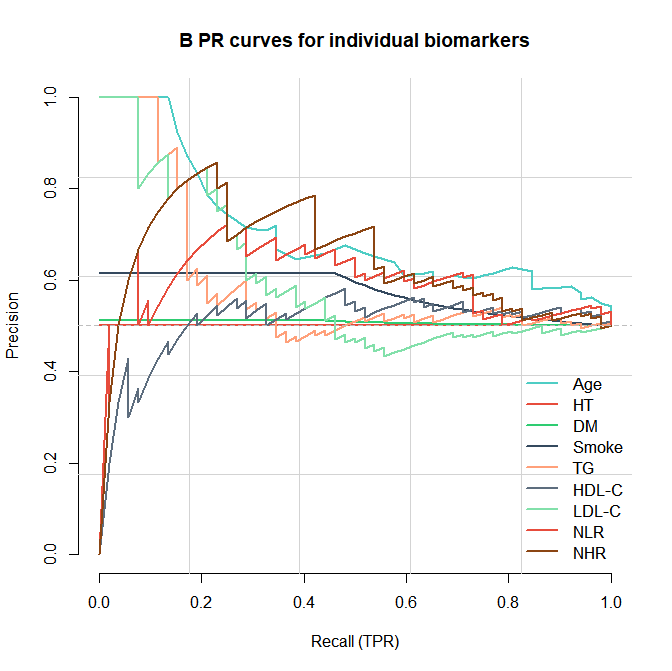
- PR 曲线
R
library(PRROC)
plot_prc_biomarkers <- function(result, title) {
n_param <- length(result)
# 严格和你原图的颜色一一对应
colors <- c(
"#4ECDC4", # Age(青)
"#E74C3C", # HT(红)
"#2ECC71", # DM(绿)
"#34495E", # Smoke(深蓝)
"#FFA07A", # TG(浅橙)
"#5D6D7E", # HDL-C(灰蓝)
"#82E0AA", # LDL-C(浅绿)
"#E74C3C", # NLR(红,和HT同色)
"#8B4513" # NHR(棕)
)[seq_len(n_param)]
# 计算每个指标 PRC
for (i in seq_along(result)) {
pr <- pr.curve(
scores.class0 = result[[i]]$scores[result[[i]]$labels == 1],
scores.class1 = result[[i]]$scores[result[[i]]$labels == 0],
curve = TRUE
)
result[[i]]$pr <- pr
}
# 画布 + 网格线(和原图一致)
plot(0:1, 0:1, type = "n",
xlab = "Recall (TPR)", ylab = "Precision",
main = title, xlim = c(0,1), ylim = c(0,1),
axes = FALSE)
# 坐标轴
axis(1, at = seq(0, 1, 0.2))
axis(2, at = seq(0, 1, 0.2))
grid(col = "lightgray", lty = "solid", nx = 5, ny = 5)
# 画PR曲线
for (i in seq_along(result)) {
lines(result[[i]]$pr$curve[,1],
result[[i]]$pr$curve[,2],
col = colors[i], lwd = 2)
}
# 画基线(和原图一样的水平虚线)
baseline <- mean(result[[1]]$labels == 1)
abline(h = baseline, lty = 2, col = "gray")
# 图例放在右下角(和原图位置一致)
legend("bottomright",
legend = names(result),
col = colors,
lwd = 2, cex = 1.1, bty = "n")
}
features <- c("age", "ht", "dm", "smoke", "tg", "hdl", "ldl", "NLR", "NHR")
names(features) <- c("Age", "HT", "DM", "Smoke", "TG", "HDL-C", "LDL-C", "NLR", "NHR")
y_true <- dat_psm$chd_num
res_pr <- list()
for (i in seq_along(features)) {
f <- features[i]
res_pr[[names(features)[i]]] <- list(
scores = dat_psm[[f]],
labels = y_true
)
}
plot_prc_biomarkers(res_pr, "B PR curves for individual biomarkers")结果展示:
- ROC曲线
R
library(pROC)
library(ggplot2)
library(dplyr)
features <- c("age", "ht", "dm", "smoke", "tg", "hdl", "ldl", "NLR", "NHR")
feature_names <- c("Age", "HT", "DM", "Smoke", "TG", "HDL-C", "LDL-C", "NLR", "NHR")
colors <- c("#4ECDC4", "#E74C3C", "#2ECC71", "#34495E", "#FFA07A", "#5D6D7E", "#82E0AA", "#E74C3C", "#8B4513")
dat <- dat_psm
dat$status <- factor(dat$chd_num, levels = c(0, 1))
roc_list <- list()
auc_vals <- c()
for (i in seq_along(features)) {
f <- features[i]
nm <- feature_names[i]
# 单因素逻辑回归
fit <- glm(status ~ get(f), data = dat, family = binomial)
pred <- predict(fit, type = "response")
# 计算ROC
roc_obj <- roc(dat$status, pred, quiet = TRUE)
roc_list[[nm]] <- roc_obj
auc_vals[i] <- auc(roc_obj)
}
legend_labels <- paste0(feature_names, " (AUC = ", sprintf("%.3f", auc_vals), ")")
ggroc(roc_list, legacy.axes = TRUE, linewidth = 1.2) +
scale_color_manual(values = colors, labels = legend_labels) +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "gray") +
labs(
x = "1-Specificity (FPR)",
y = "Sensitivity (TPR)"
) +
theme_bw() +
theme(
panel.grid.major = element_line(color = "lightgray", linetype = "solid"),
legend.position = c(0.80, 0.20), # 图例位置和原图一致
legend.title = element_blank(),
legend.background = element_rect(fill = "white", color = NA)
)结果展示:
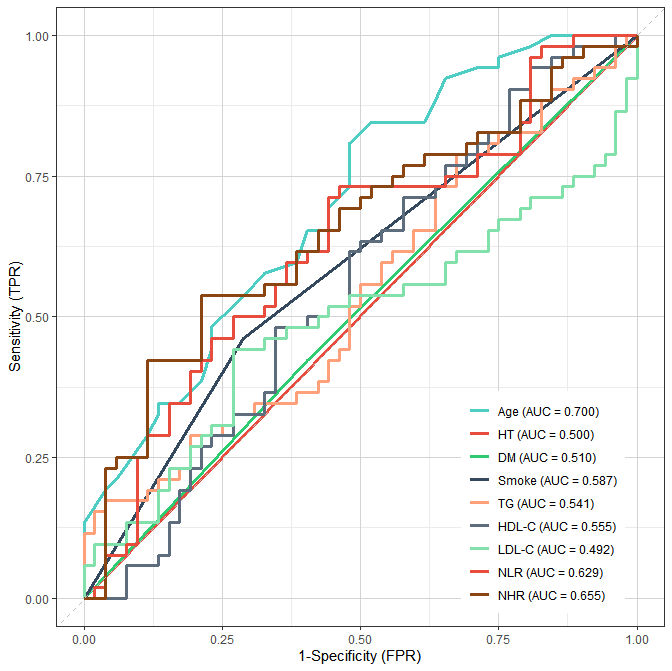
- 校准曲线
R
library(ggplot2)
# 1. 构建数据(绝对不报错)
cal_df <- data.frame(
predy = cal[, "predy"],
apparent = cal[, "calibrated.orig"],
corrected = cal[, "calibrated.corrected"]
)
# 2. 【最终完整版】校准曲线 + 自动图例
ggplot(cal_df) +
# 理想线
geom_abline(aes(color = "Ideal line"), intercept = 0, slope = 1, linetype = "dashed", linewidth = 1) +
# 两条校准线
geom_line(aes(x = predy, y = apparent, color = "Apparent"), linewidth = 1.2) +
geom_line(aes(x = predy, y = corrected, color = "Bias corrected"), linewidth = 1.2) +
# 颜色严格匹配你要的
scale_color_manual(
name = NULL,
values = c(
"Apparent" = "#4ECDC4",
"Bias corrected" = "#E74C3C",
"Ideal line" = "gray"
)
) +
# 坐标轴
labs(x = "Predicted Probability", y = "Observed Probability") +
xlim(0, 1) + ylim(0, 1) +
# 主题 + 图例位置
theme_bw() +
theme(
panel.grid = element_blank(),
legend.position = c(0.85, 0.05), # 右下角
legend.background = element_rect(fill = "white", color = NA)
)结果展示:
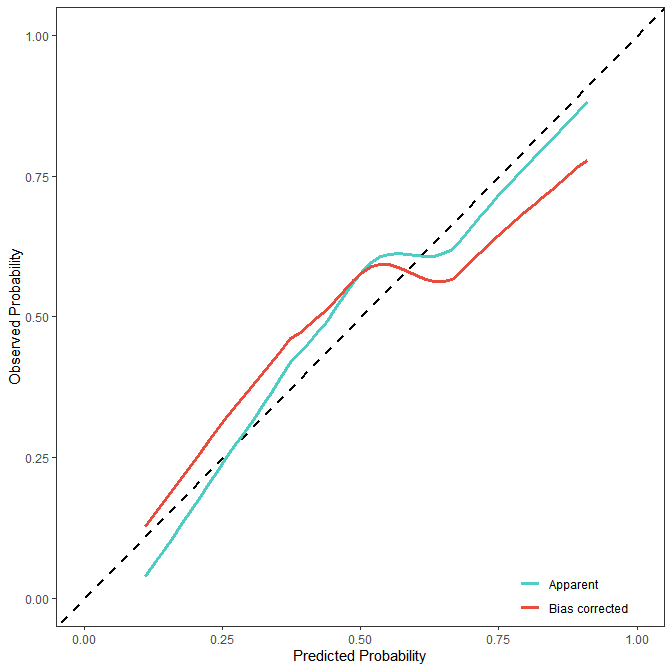
- ROC曲线
R
library(ggplot2)
library(pROC)
# 计算 ROC、AUC、CI
pred_multi <- predict(fit, type = "fitted")
roc_obj <- roc(dat_psm$chd_num ~ pred_multi, quiet = TRUE)
auc <- round(auc(roc_obj), 3)
ci <- round(ci.auc(roc_obj), 3)
# 图例文字(自动换行)
legend_label <- paste0("Model\nAUC = ", auc,"\n95%CI: ",ci[1],"-",ci[3])
# ✅ 最终版:ggroc + 右对齐 + 黑色文字 + 无网格
ggroc(roc_obj, legacy.axes = TRUE, color = "#4ECDC4", linewidth = 1.2) +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "gray", linewidth = 1) +
# 右对齐 + 黑色文字
annotate("text", x = 0.99, y = 0.05, hjust = 1, size = 4.2, color = "black", label = legend_label) +
labs(x = "1 - Specificity", y = "Sensitivity") +
xlim(0, 1) + ylim(0, 1) +
theme_bw() +
theme(panel.grid = element_blank(), legend.position = "none")结果展示:
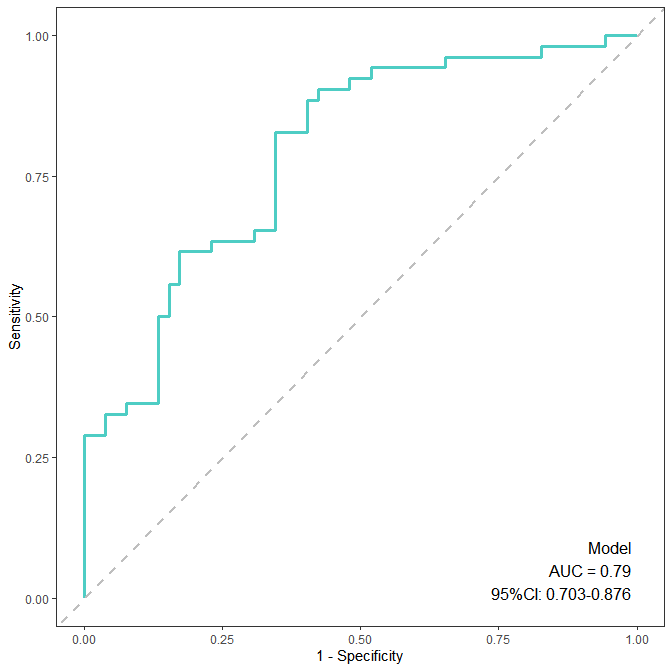
四、小结
本章以 NAFLD 合并 CHD 共病风险研究为切入点,依托模拟临床数据集,完整复现了当下高分临床共病研究的标准化分析全流程,串联起传统临床统计、混杂因素校正、非线性关系挖掘与可解释机器学习的全套研究范式,实现从变量筛选、模型构建到结果可视化验证的闭环落地。
首先,研究基于真实临床分布规律构建模拟数据,纳入人口学、基础生化、血常规等核心指标,批量计算 SII、SIRI、NHR、NLR 等 10 项临床常用免疫炎症复合标志物,完整还原真实回顾性研究的数据结构与变量体系。在此基础上,通过基线特征统计完成两组人群临床资料的组间对比,清晰反映 NAFLD 单纯组与合并 CHD 共病组在炎症、代谢指标上的基线差异,为后续分析奠定基础。
在传统统计层面,先通过单因素与多因素 Logistic 回归筛选共病发生的独立危险因素,明确 NHR、NLR、SIRI、PNR 等炎症指标的临床预测价值,并结合森林图直观展示各指标的效应量与置信区间;同时采用 LASSO 回归压缩降维、筛选核心混杂变量,搭配 1:1 倾向得分匹配(PSM)平衡组间混杂偏倚,最大程度降低回顾性研究的选择偏倚,提升结论的可靠性。
针对临床指标非线性关联的研究需求,本研究利用限制性立方样条(RCS)模型,探究各炎症标志物与 CHD 发病风险的线性 / 非线性剂量反应关系;结合 Spearman 相关性分析,进一步验证炎症指标与 Gensini 冠脉病变严重程度评分的关联强度,从 "发病风险" 与 "疾病严重程度" 双维度,阐明免疫炎症紊乱在共病进展中的作用。
在进阶建模与人工智能解读部分,统一构建 GLM、SVM、随机森林三种预测模型,通过残差分布对比优选模型;依托 SHAP 可解释分析,从特征贡献度、分组差异、单样本归因多角度拆解模型机制,规避传统黑箱 AI 难以临床解读的短板。同时结合列线图、ROC 曲线、PR 曲线、校准曲线多维度评价模型区分度、精准度与拟合效果,形成一套规范、完整的预测模型验证体系。
到这里,本篇 NAFLD+CHD 共病 + 炎症标志物 + 可解释机器学习高分 SCI 完整复刻就全部结束了,整套流程从数据构建、统计分析、非线性探索,到机器学习建模、SHAP 结果解读、模型多维度验证一步不落、全套打通,完全复刻顶刊研究标准范式;后续想看哪些方向的论文复刻,比如代谢共病、肿瘤预后、影像组学、列线图预测模型、免疫评分、孟德尔随机化等选题,都可以在评论区留言,点赞收藏越多更新速度越快,我也会持续拆解高分 SCI 研究套路,分享可直接套用的全套科研代码与分析流程。