
最近在一对一辅导一位同学做全基因组关联分析(GWAS)项目。他在处理一批水稻的数据时,使用 rMVP 跑流程遇到了几个非常典型的坑。从上千万个原始变异位点的质控过滤,到代码报错排查,再到最终模型选择,在这里记录总结下,供开展植物 GWAS 的同行参考。
一、 过滤位点过犹不及
在使用 PLINK 对原始 VCF 文件(通常包含上千万个位点)进行过滤时,很多人会直接套用人类代码,加上 --hwe 1e-6 参数。但在植物群体(尤其是水稻等高度纯合的群体)中,这会导致灾难性的后果------极高比例的真实高质量位点被当作测序错误误删。
**原因分析:**哈迪-温伯格平衡(HWE)的前提是群体随机交配,期望存在一定比例的杂合子。而自交系作物的纯合度接近 100%,极度缺乏杂合子。这种天然的群体结构在算法看来是对 HWE 的严重偏离,从而产生极小的 P 值触发过滤机制。
**建议:**植物纯系 PLINK 过滤脚本不加 --hwe 参数。
bash
plink --vcf raw_data.vcf.gz \
--keep-allele-order \
--snps-only just-acgt \
--biallelic-only strict \
--maf 0.05 \
--geno 0.1 \
--chr 1-12 \
--allow-extra-chr \
--make-bed \
--out rice_filtered_clean注:保留 --chr 1-12 可以剔除未挂载的 scaffold(如 ChrUn, ChrSy),这些非数字染色体编号会导致 rMVP 后续画图直接崩溃。
二、 跑项目时,先用小数据测通
项目数据较大,无论是在开发还是跑项目,尽量用小数据集测试,先跑通再说,这是基本常识。
提取测试数据:
bash
plink --bfile rice_filtered_clean --thin 0.01 --make-bed --out rice_test建议:在将完整脚本提交到集群(如 SLURM)正式运行前,可用 1% 的数据进行测试,排查环境与代码报错。
三、 关于GWAS模型选择
GWAS软件很多,在这里只针对他的项目,用rMVP软件。
rMVP 原生支持三种主流模型,如果是做自己的项目,建议使用 method = c("GLM", "MLM", "FarmCPU") 同时跑出三种结果进行比对。
-
GLM (一般线性模型): 仅校正群体结构(PCA)。计算极快,但忽略了家系间的亲缘关系(Kinship),容易产生大量假阳性。
-
MLM (混合线性模型): 同时纳入 PCA 和 Kinship,是经典的Q+K模型。能极大程度压制假阳性,但缺点是存在过度拟合风险,容易把真实的微效信号抹杀掉(假阴性高)。
-
FarmCPU: 采用多基因座(Multi-locus)模型,在固定效应和随机效应之间循环迭代。它的统计功效(Power)最高,背景噪音压得极低,而真实信号极度锐利。
rMVP更新比较频繁,有些参数可能会发生变化,注意版本。
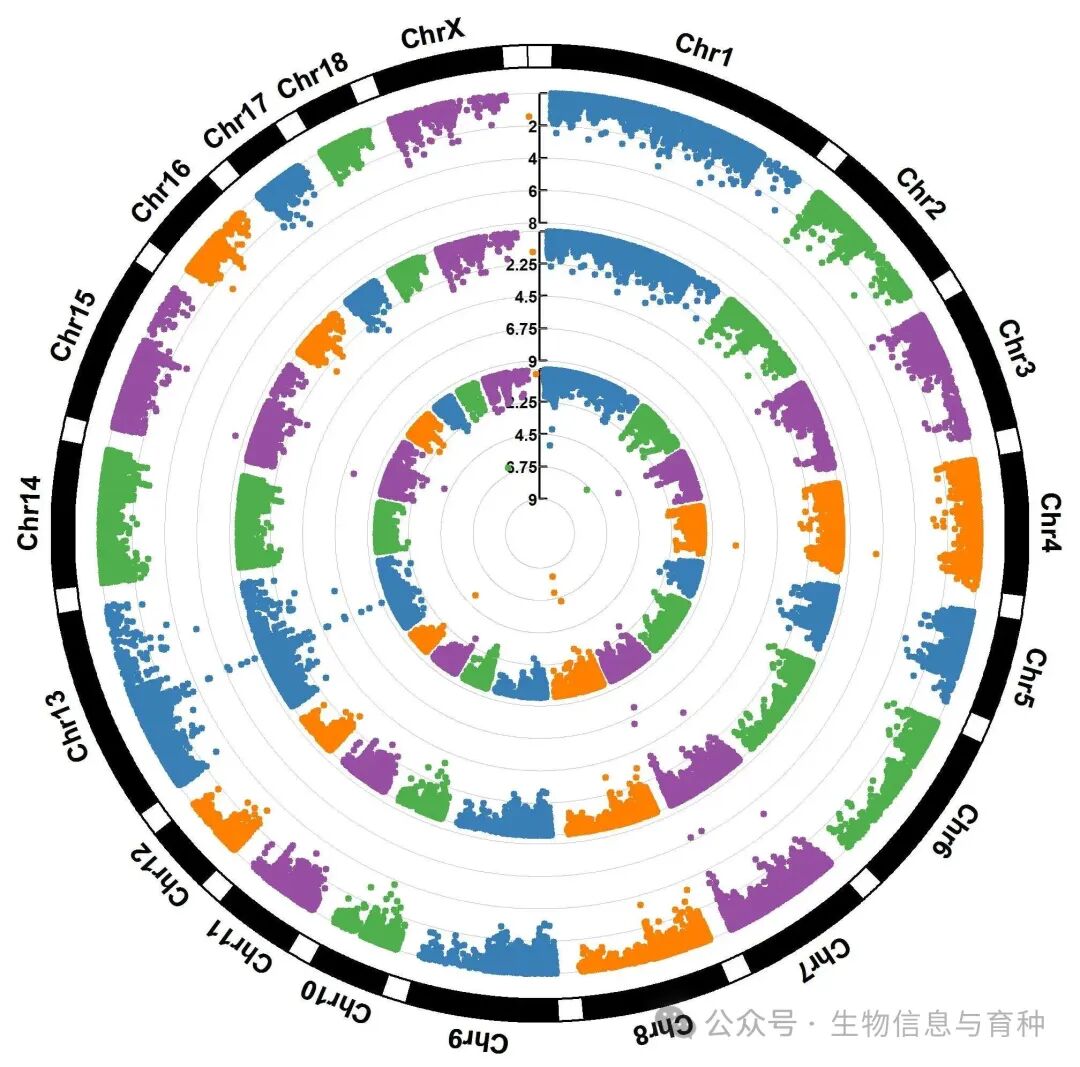
**建议:**如果要发文章,建议以 FarmCPU 的结果为主 进行靶区挖掘,同时将 MLM 的结果作为交叉验证。如果一个位点在 FarmCPU 和 MLM 中同时显著,该区间即为超高优先级的候选靶点。
四、 GWAS 需要同时用 SNP 和 InDel 吗?
虽然 InDel往往是导致表型变异的直接原因(如移码突变),但在全基因组关联扫描阶段,只使用高质量的 SNP 即可,不建议合并或单独运行 InDel。
-
分型准确率考量: 二代测序对 InDel 的 calling 错误率远高于 SNP,合并运行容易污染底层的 PCA 和 Kinship 矩阵,降低统计功效。
-
连锁不平衡(LD)效应: 高密度的 SNP 标记(如过滤后依然保留 200~300 万个)足以作为任何真实 InDel 的标签。
建议: GWAS 阶段用纯 SNP 数据,锁定显著候选区间后,再回到原始 VCF 文件中,将该区间内的 SNP 和 InDel 同时提取出来,进行单倍型分析与功能候选基因的比对。
[