文章目录
从芯片到NGS高通量测序,从bulk到single cell的RNA-seq,只要是涉及到gene对象,一个必不可少的分析项目,就是富集分析。
不管是转录组,还是芯片数据,或者其他有关基因的组学分析,每当数据分析到后面,要想得到结果,都躲不过这个富集分析,因为它是帮助我们从庞杂的组学数据中发掘规律重要的一环。对基因功能进行富集分析, 就有可能发现在生物学过程中起关键作用的生物通路, 并且帮助理解生物学过程的分子机制。
1,定义
基因富集分析 是在一组基因中找到具有一定基因功能特征和生物过程的基因集,在研究差异表达基因、筛选基因的后续分析中经常使用。
其实定义中已经明确了,一组gene(就是我们的背景gene)、一定功能特征和生物过程(富集结果)、找到的gene集(一般是我们想要富集分析想要挖掘功能的gene对象)。
基因集,也叫gene set,也就是一系列具有相同功能的基因构成的集合,比如某一条代谢通路(pathway),其中有很多的基因,因此位于同一条通路下的基因就构成了一个基因集合。 组成基因集的最基本元素就是一个一个的基因。
参考:https://www.jieandze1314.com/post/cnposts/28/


2,富集分析算法

参考上述文献,大体上富集分析有四类算法:ORA(过表征分析,over-representation analysis, ORA)、FCS(功能集打分,functional class scoring, FCS)、PT(基于通路拓扑结构,pathway topology, PT)、NT(基于网络拓扑结构,network topology, NT);
核心就在于下图:
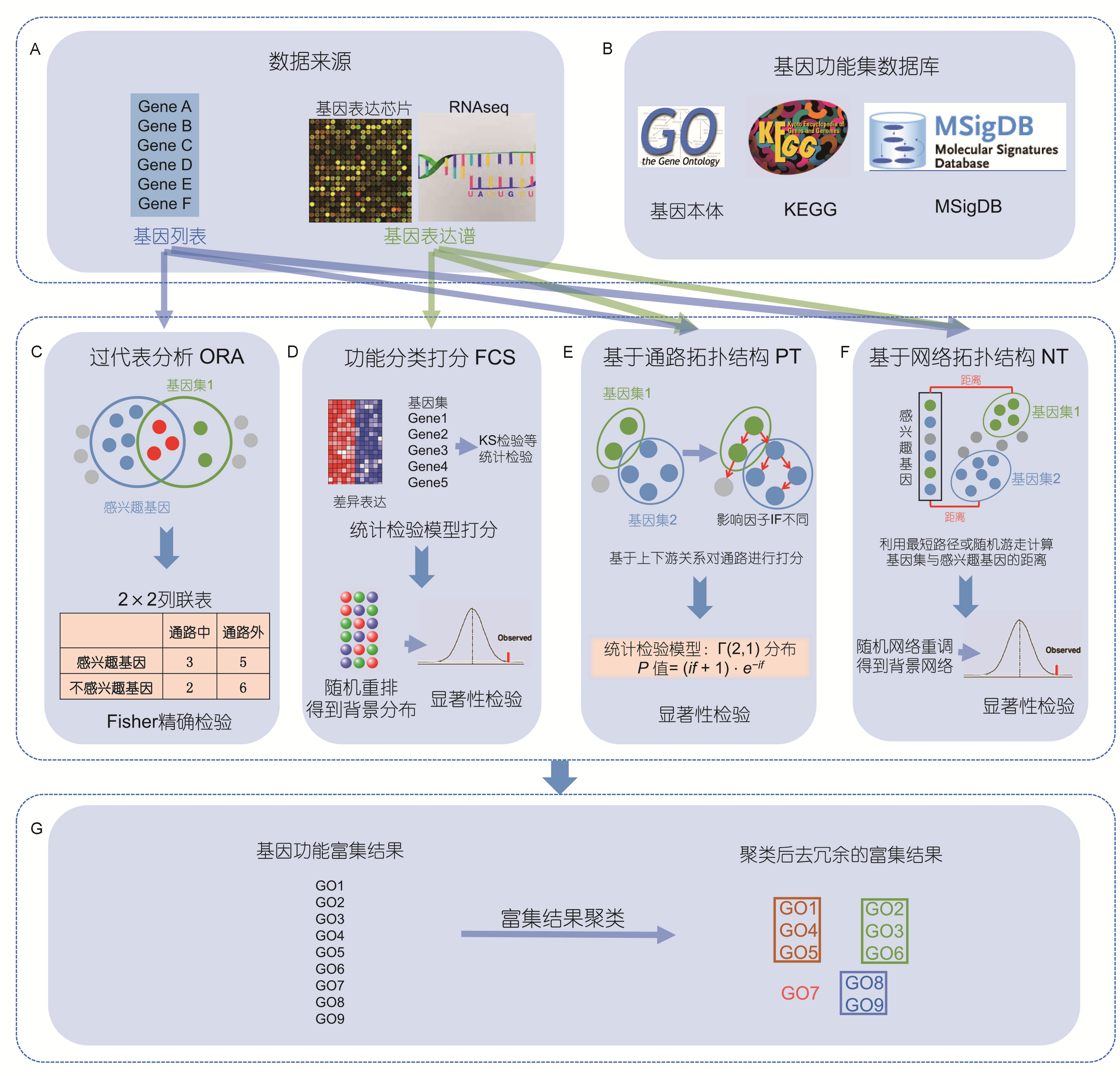
我们一般分析RNA-seq数据的时候(bulk或sc),其实经常用到的无非是ORA(超几何分布model,做Fisher精确检验的那个),以及GSEA(基于rank 秩的分析,也就是所谓的Gene Set enrichment analysis);
其实富集分析的同义说法/表述现在有很多(基本上你去找文献,method里面只要是enrichment的,什么新词都能看到),什么GO富集分析、KEGG富集分析、基因集富集分析(就是上面GSEA)、功能富集分析、功能分析、通路分析,总之,一般门外汉或者是刚入门生信分析的新手常常会把这些一大堆词挂在口边(有很多新词其实就是那些门外汉生造出来的),又经常分不开哪个是哪个,用的是什么算法,基本上都是clusterprofiler 等R包一用、代码一抄,也就不管三七二十一了,这是极不负责任的生信学习方式!
简单来说,ORA是目前应用最多的方法,GO富集分析和KEGG富集分析就是使用的这种方法;FCS这种方法应用于GSEA分析(clusterProfiler就是)。

(1)基因功能富集分析的基因功能数据库和数据类型
基因功能富集分析中的基因功能指的是众多代表一定的基因功能特征和生物过程的基因功能集(gene set)。 由这些基因功能集构成的常用基因功能数据库有GO, 生物学通路, 包含生化反应、代谢或信号通路的KEGG, Reactome, Biocarta等, 整合数据库, 如MsigDB等。
在功能组学研究中, 研究者通常会获得一组他们感兴趣的基因, 如在疾病和正常组织中有显著差异表达的基因, 在药物或外界环境刺激下特定组织中表达水平有显著异常的应激基因等. 要揭示其中隐含的生物学分子机制, 研究者可针对这组感兴趣的基因, 进行基因功能的富集分析, 发现在其中有显著富集的特定生物学通路, 从而从分子机制上来解释所观察到的生物学现象. 除此以外, 高通量组学技术, 如基因表达芯片(microarray)或RNA-seq, 可获得基因组中所有基因的表达水平. 为充分利用获得的高通量数据, 研究者也可以直接针对全基因组基因表达谱信息来进行富集分析, 从中鉴定出案例和对照状态下在研究对象中发生显著表达差异的生物通路, 从而揭示其中的生物学分子机制. 针对这些不同的数据需要开发不同的功能富集分析方法. 例如, 对于基因表达芯片和RNA-seq, 在富集分析过程中原始数据的处理方式是不同的. 其中, 基因芯片记录的是连续的荧光信号强度值, 而RNA-seq记录的是RNA序列的读段个数, 需要采用不同的统计模型进行分析。
(2)ORA过表征分析
算法原理
ORA针对的数据是一组感兴趣的基因(基因列表), 其目的是在这组基因中发现有明显统计学上富集的基因功能集。
ORA的目的,在于判断某一类功能或分类是否在特定基因集中显著富集。
其基本步骤包括先将给定的基因列表与待测功能集做交集, 找出其中共同的基因并进行计数(统计值), 最后利用统计检验的方式来评估观察的计数值是否显著高于随机, 即待测功能集在基因列表中是否显著富集。
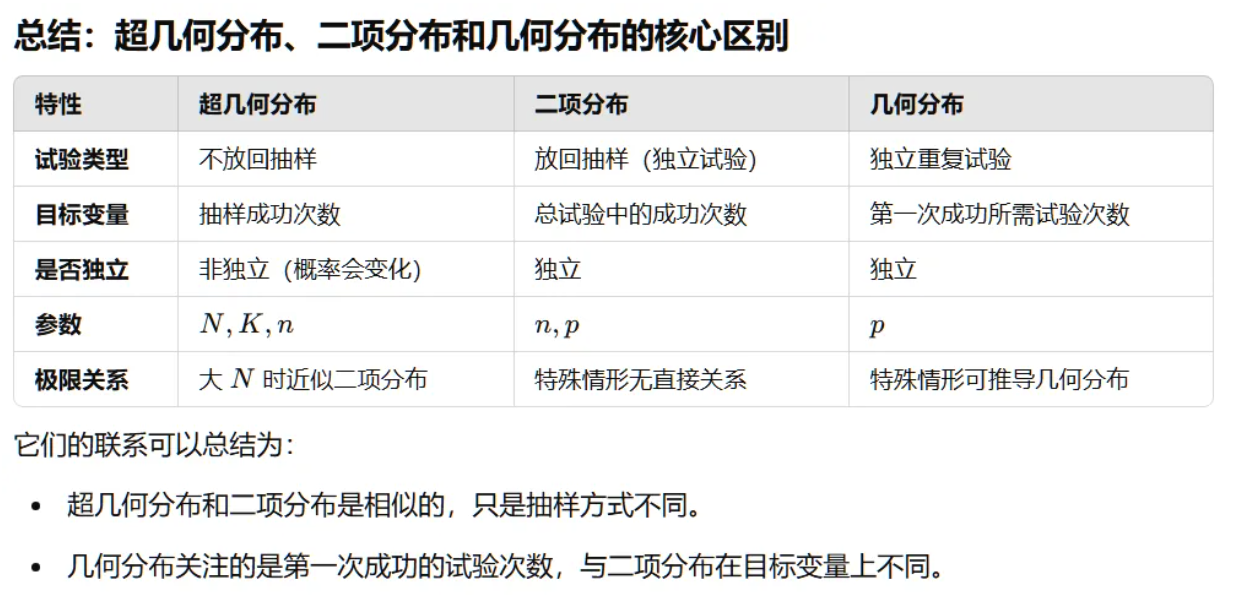
常见的统计学方法有卡方检验, Fisher精确检验和二项分布检验, 而其中最为广泛使用的是Fisher精确检验, 即利用2×2的列联表, 根据超几何分布来检验基因列表中的基因在待测功能集中是否显著富集。
超几何分布被用于评估特定功能基因集在差异表达基因列表中的过表达程度,而 Fisher 精确检验(Fisher's Exact Test)正是基于超几何分布计算 p 值。
超几何分布是本科生学概率统计的时候都应该知道的最基础的知识:


实际上,超几何分布中总体只有参数N(总个数)、K(不及格的个数,或者是成功/不成功的类别个数),然后就是我们抽样的样本数n,
然后这个离散分布,计算的其实是我们抽样n中成功类别k个的概率值(成功类别k,或者是不及格个数k,总之我们的目标变量是抽样成功的次数)
ORA的目标是判断在一个感兴趣的基因集(例如差异表达基因)中,某个特定功能类别(如 KEGG通路)是否显著富集。
注意下面的H0零假设,以及H1备择假设:

注意:这里我们要找到的是X≥k的累计概率, 直接反映了观察结果及更极端情况的可能性, 如果 p-值很小(如 <0.05),说明观察值极难由随机分布产生,因此可以拒绝零假设,认为有富集现象。
简单举个例子:



下面举一个实际例子来分析:
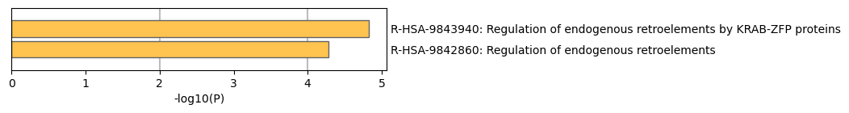
比如说我们human,正常的所有蛋白质gene(大概)在2w5k个gene左右,25k;
然后比如说以上面图中的这个通路"内源逆转录元件调控(第2项)"为例,
假设这个通路包含了人类25k gene中的5k gene,
即人类所有蛋白质编码的25k gene中,5k在这个通路中,20k不在这个通路中;
然后现在我们手头上有一个感兴趣的研究gene集,比如说大概在1k个左右,
然后正好发现,这1k个gene中有0.3k是在这个通路中,另外0.7k不在这个通路中;
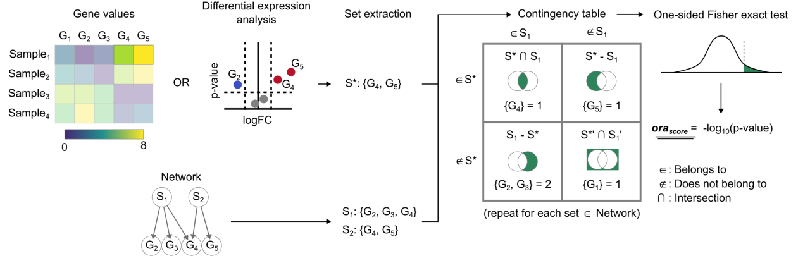
这其实就已经有了一个2x2的列联表了:
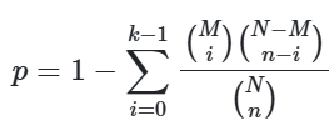
这个图片其实就很明显了,
2x2的列联表,首先s是我们感兴趣的gene list,第1行是∈s,第2行∉s*;
然后就是我们domain knowledge中关于某个通路的gene集,第1列是属于某个通路的∈s1,第2列是∉该通路的所有gene。
是否在我们感兴趣的gene列表中,与是否在某个通路中,这两个因素之间是否存在某种关系?
在这个等式中,**N是背景分布中基因的总数(背景gene总数),M是该分布中被注释到感兴趣通路(基因集)的基因的数量,**n是(我们手头上有的)感兴趣基因列表的大小,k是该列表中被注释到感兴趣通路(基因集)的基因的数量。默认情况下,背景分布是所有带有注释的基因。p值应调整为多次比较。
两个小问题:为什么是取比观察值更极端的值的概率之和,以及为什么是多重假设检验?
第1个问题:比如说前面给出的这个图,为什么是观察到10个(当前的抽样情况)或更多个显著gene,为什么是≥10?
这个其实也是本科生概率统计的基本功,假设检验中p值的定义,实际上就是计算在原假设H0成立的情况下,观察到的数据或者是更极端的数据出现的概率,通过计算比当前观察值k更极端的取值的概率之和,可以量化当前观察值的显著性。如果p值小于某个阈值(如0.05),则认为目标基因集在该功能类别中显著富集。
再往下说,其实就是假设检验的基本原理,就是小概率事件原理,我们拒绝H0原假设,其实是基于反证法的逻辑:
如果H0成立,那么我们就能够观察到基于H0假设的事件,如果我们能够观察到一件事,其实基于极大似然原理,我们计算出来统计量取当前观察值的概率就不会是一个非常小的值,就应该是一个可观的取值;
反之,如果我们的统计量,计算当前观察到的值或更极端的值出现的概率都是一个非常小的值,那只能说明(依据随机变量的值-事件的逆映射)基于H0假设的事情是一个小概率事件,也就是即使满足H0假设,但是这个事件却基本不可能发生(与满足H0假设的本质矛盾),所以我们才说H0是错误的,当然也不能说绝对的拒绝,至少我们没有足够的证据接受H0。
那么,为什么要+更极端的取值呢?长话短说,这是由于H1也就是备择假设决定的极端方向;
python
1. 关键逻辑重构:H₁决定"极端"的方向
(1) 假设检验的本质是"比较"
● H₀(零假设):代表"无效应"或"随机"的基准模型(如"基因无富集")。
● H₁(备择假设):代表你真正关心的科学假设(如"基因显著富集")。
(2) H₁的定义决定了P值的计算方式
● H₁的方向性分为两种:
○ 单侧H₁(如"富集更多"):只关心一个方向的偏离。
○ 双侧H₁(如"富集更多或更少"):关心两个方向的偏离。
● P值必须与H₁一致:
○ 若H₁是"富集更多",P值=(P(X≥x))(右尾概率)。
○ 若H₁是"富集更少",P值=(P(X≤x))(左尾概率)。
○ 若H₁是"富集差异"(无方向),P值=(P(X≥x) + P(X≤对称点))(双尾概率)。
(3) 为什么不能只用单点概率?
● 单点概率无法体现H₁的方向性:
例如,观察10个基因富集(期望5个):
○ 若H₁是"富集更多",需计算(P(X≥10))(包括10、11、12...)。
○ 若H₁是"富集更少",需计算(P(X≤10))(包括0、1、2...10)。
○ 单点(P(X=10))对两者相同,无法区分方向!
2. 通过例子彻底理解
例子1:药物有效性检验
● H₀:药物无效(治愈率=50%)。
● H₁:药物有效(治愈率>50%)。
● 数据:试验10人,治愈8人。
分析:
● 单点概率:(P(X=8)=\binom{10}{8}0.58 0.52 \approx 0.044)。
● 累积概率(右尾):(P(X≥8)=P(8)+P(9)+P(10)\approx 0.054)。
问题:若用单点概率0.044(<0.05),会得出"显著"结论;但累积概率0.054(>0.05)不显著。
关键:单点概率忽略了"更极端情况"(如9人或10人治愈),而H₁明确指向"更多治愈",必须累加右尾!
例子2:基因富集分析(我们的案例)
● H₀:目标基因集与通路无关(随机分布)。
● H₁:目标基因集在通路中富集更多(科学假设)。
● 数据:100个基因中15个属于通路(期望10个)。
P值计算:
● 右尾累积概率:(P(X≥15)=\sum_{k=15}^{100} P(X=k))。
○ 包含15、16、...100的所有"比观察值更富集"的情况。
● 若错误地用单点概率:
○ (P(X=15))可能很小,但忽略了"16个基因更支持H₁"的证据!
3. 为什么说"更极端值"是加强命题?
● 科学假设需要"一致性证据":
H₁声称"存在效应",而效应越强(如富集基因数越多),越支持H₁。因此,P值需累积所有至少同样支持H₁的证据。
● 类比法律审判:
○ 若被告涉嫌犯罪,法官不仅考虑"恰好找到一件指纹",还需综合"指纹、DNA、监控"等所有同等或更强证据。
○ 单点概率类似"仅凭指纹定罪",而累积概率是"综合所有证据"。
4. H₁如何从科学问题转化为统计假设?
步骤1:明确科学目标
● 例如:"有IDR的锌指蛋白是否在发育通路中显著富集?"
○ 这里H₁的方向是"富集更多"(单侧)。
步骤2:选择对应的统计检验
● 超几何检验的右尾P值((P(X≥x)))直接对应这一假设。
步骤3:拒绝H₀的条件
● 若(P(X≥x)<α)(如0.05),说明在"富集更多"方向上,数据极不支持H₀。
5. 如果你坚持用单点概率会怎样?
● 问题1:方向性丢失
单点概率无法区分"富集更多"还是"富集更少"。例如:
○ (P(X=15))和(P(X=5))在单点概率下可能相同,但生物学意义相反。
● 问题2:决策不稳定
离散分布中,单点概率随观察值微小变化剧烈跳跃(如14→15基因时P值突变),而累积概率更平滑。
● 问题3:违背科学逻辑
科学假设通常关注"趋势"(如"越多富集越支持H₁"),而非精确值。
6. 终极答案:为什么必须累加更极端值?
1. H₁的方向性要求:科学假设明确指向"富集更多"或"更少",P值必须与之匹配。
2. 证据的累积性:比观察值更极端的数据同样(甚至更强)支持H₁,忽略它们会低估证据强度。
3. 稳定性与控制错误率:累积概率避免单点概率的跳跃问题,确保统计结论的稳健性。
总结
● H₁的定义决定了P值的计算方式(单尾或双尾)。
● "更极端值"的累加是为了全面量化数据对H₁的支持程度,而不仅是"恰好观察值"的偶然性。
● 单点概率无法满足科学假设的定向需求,会导致逻辑矛盾或决策不稳定。
直觉上讲------"加强命题"是对的!累加更极端值本质上是将所有至少同样支持H₁的证据纳入统计决策,从而更严谨地回答科学问题。如果不看上面那一大堆废话的话,其实也可以从其他角度来理解:
单点概率计算,离散分布很简单,能够算;但是连续的就不能够计算了,所以连续分布需要一个区间,而不是一个单点取值。
python
1. 极大似然原理与假设检验
极大似然原理是统计学中的一个基本原理,它指出在给定一组数据时,
最有可能产生这组数据的参数值是使似然函数最大化的参数值。
在假设检验中,我们通常假设原假设((H_0))成立,
然后计算在这一假设下观察到当前数据或更极端数据的概率。
2. 观察到的事件与小概率原理
如果原假设((H_0))成立,那么基于这一假设的事件(即观察到的数据)应该是合理的,
即其出现的概率不应该是一个非常小的值。
如果观察到的事件本身就是一个小概率事件,那么根据小概率原理,我们有理由拒绝原假设。
3. 计算更极端的值
我们计算当前观察到的值或更极端的值出现的概率,是因为如果连这个合事件的概率都非常小,
那么单个取值的事件的概率也会更小。这实际上是一种"加强命题"的方法,即如果在原假设下,
观察到的数据或更极端的数据出现的概率非常小,那么我们更有理由拒绝原假设。
4. 假设检验的决策
在假设检验中,我们通常设定一个显著性水平(如0.05),
如果计算出的p值小于这个显著性水平,我们就拒绝原假设。
这并不意味着我们绝对地拒绝原假设,而是说在给定的显著性水平下,
我们没有足够的证据接受原假设。
python
1. 逻辑链总结
1. H₀成立时:观察到的统计量(或更极端值)应是一个较大概率事件(即P值较大)。
2. 若P值很小:说明在H₀下,当前观察值(及更极端情况)几乎不可能发生,与H₀的预期矛盾。
3. 统计决策:小P值表明数据不支持H₀,但无法"证明"H₀绝对错误(只能拒绝或不拒绝)。
2. 关键点强化
(1) 为什么必须包含"更极端值"的概率?
● "极端性"定义需一致:假设检验的P值必须预先定义"极端方向"(由H₁决定)。例如:
○ 富集分析(右尾检验):关心"是否比随机更多",因此P值=(P(X≥x))。
○ 缺失分析(左尾检验):关心"是否比随机更少",P值=(P(X≤x))。
● 单点概率无法体现方向性:若仅用(P(X=x)),无法区分富集(右尾)或缺失(左尾)。
(2) 极大似然与假设检验的关系
● 极大似然估计(MLE):寻找使观察数据概率最大的参数(如"骰子是否公平")。
● 假设检验:固定H₀的参数(如"骰子公平"),计算数据与此假设的兼容性。
○ 若数据似然在H₀下极低,说明H₀可能不合理。
○ 但假设检验比MLE多了一步:量化"极端性"以控制错误率。
(3) "矛盾"的统计学解释
● 小P值的含义:
○ 在H₀下,当前数据(及更极端数据)的总体概率很小(如P=0.01)。
○ 这并非"H₀绝对错误",而是"数据与H₀的兼容性极低"。
○ 统计学中称之为"显著性"(significance),而非"确定性"。第2个问题:为什么过表征分析是多重假设检验?
到底是哪里在对多个假设进行检验?
过表征分析的核心是判断某一功能类别(如GO term或KEGG通路)中的基因是否在目标基因集中显著富集。具体来说,对于每一个功能类别,都会进行一次假设检验。假设检验的基本步骤如下:
- 原假设(H0):目标基因集在该功能类别中没有富集。
- 备择假设(H1):目标基因集在该功能类别中有富集。
在实际分析中,通常会针对多个功能类别(如成百上千个GO term或KEGG通路)分别进行假设检验。例如,如果有1000个功能类别,就需要进行1000次假设检验。这种同时对多个假设进行检验的情况称为多重假设检验。
多重假设检验的一个重要问题是假阳性率的增加。如果对每个假设单独进行检验,假阳性率(即错误拒绝原假设的概率)为(α)(通常取0.05)。但在多重假设检验中,假阳性率会显著增加。例如,对于1000次假设检验,即使每次检验的假阳性率为0.05,整体假阳性率会远高于0.05。因此,需要对p值进行校正,以控制假阳性率。
常见的校正方法包括:
- Bonferroni校正:将每个p值乘以假设检验的次数(如1000),从而严格控制整体假阳性率。
- Benjamini-Hochberg校正(FDR校正):控制假发现率(False Discovery Rate, FDR),允许一定比例的假阳性,但整体假阳性率不会过高。
再次强调,gene集的多重假设检验是针对通路进行的(有多少个通路,就进行多少次假设检验),不是针对gene的(我们的gene集是作为一个整体来参与假设检验的)!!!
python
解释:
1. 背景:
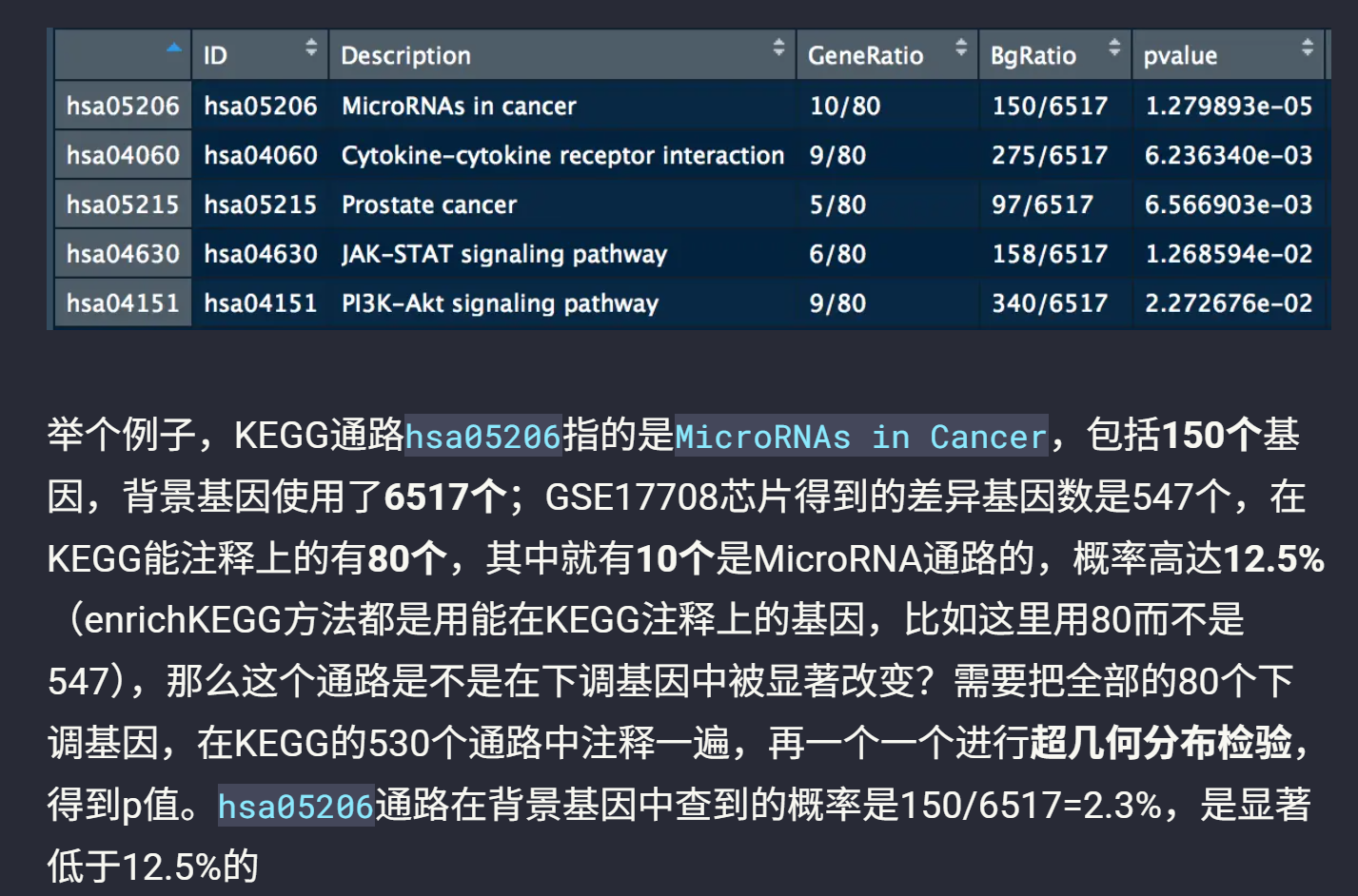
○ 文章中提到有80个下调基因,这些基因需要在KEGG数据库的530个通路中进行注释。
○ 每个通路是一个独立的假设,假设这些基因是否在某个通路中显著富集。
2. 多重假设检验的对象:
○ 针对通路:每个通路是一个独立的假设,需要检验这些基因是否在该通路中显著富集。因此,多重假设检验是针对这530个通路进行的。
○ 不是针对基因:虽然有80个基因,但这些基因是作为一个整体来检验它们在每个通路中的富集情况,而不是对每个基因单独进行假设检验。
具体过程:
● 注释:将80个下调基因在KEGG的530个通路中进行注释,看这些基因是否属于某个通路。
● 超几何分布检验:对每个通路进行超几何分布检验,计算p值,判断这些基因在该通路中是否显著富集。
● 多重假设检验:因为有530个通路,每个通路的检验都是一个独立的假设,因此需要进行多重假设检验来校正p值,避免假阳性。
总结:
这里的多重假设检验是针对通路进行的,而不是针对单个基因。其实这个问题还是挺基础的,也比较典型,我也在面试的时候问过一个考研学生相关问题,就是一个很简单的问题,通过这个问题,我就能够知道这个学生到底做没做过基本的生物信息学组学数据分析,如果做过了到底具不具备基本的统计学常识,到底是真懂还是假懂,其实我看他的回答就能知道,有人一问就漏出了马脚。
问题如下:
假设你在进行RNA-seq差异表达分析后,得到了1000个差异基因。你决定使用ORA(Over Representation Analysis)方法来分析这些基因是否在某个特定的生物学通路中显著富集。已知:
- 人类基因组中共有25,000个基因(背景基因)。
- 在DNA修复通路(DNA repair pathway)中,共有1,500个基因。
- 你发现这1000个差异基因中有300个基因与DNA修复通路的基因集合有交集。
- 你总共要分析30个不同的通路。
问题:
- 在这种情况下,你需要进行多少次假设检验?(或者另外一种问法:你是否需要进行多次假设检验,如果是的话,你需要进行多少次假设检验?以及你是否需要对多重假设检验进行矫正,如果是的话,你打算如何进行矫正)
- 选项:
- A. 1000次(针对每个差异基因)
- B. 300次(针对DNA修复通路中的每个基因)
- C. 25,000次(针对所有背景基因)
- D. 1,500次(针对DNA修复通路中的所有基因)
- E. 30次(针对每个通路)
- 选项:
- 解释你的选择 :
- 为什么你选择这个选项?请详细说明你的理由。
- 多重假设检验的必要性 :
- 如果你需要进行多重假设检验,那么选择哪种校正方法更适合?请解释其原理和适用场景。
- 如果不需要,为什么?
参考答案:
- 假设检验的次数 :
- 正确答案是 E. 30次。
- 解释:在ORA分析中,每次假设检验的目的是判断一组差异基因是否在某个特定通路中显著富集。因此,每次检验是针对一个通路进行的,而不是针对单个基因。因为你有30个通路需要分析,所以需要进行30次假设检验。
- 多重假设检验的必要性 :
- 需要进行多重假设检验。因为你在同时对30个不同的通路进行检验,每个通路的检验都是一个独立的假设。如果不进行多重假设检验,可能会增加假阳性的风险。
- 校正方法 :
- Bonferroni校正:将每个p值乘以假设检验的次数(30)。这种方法非常保守,适用于对假阳性要求非常严格的场景。
- Benjamini-Hochberg校正:控制假发现率(FDR),适用于大规模假设检验,能够更好地平衡假阳性和假阴性。
- 讨论 :
- 不进行多重假设检验的问题:如果不进行多重假设检验,可能会导致大量的假阳性结果,即错误地认为某些通路显著富集,而实际上它们并不是。
- 选择合适的校正方法:在实际研究中,选择校正方法需要根据研究的具体需求。如果对假阳性非常敏感,可以选择Bonferroni校正;如果需要在假阳性和假阴性之间取得平衡,可以选择Benjamini-Hochberg校正。
再补充一点吧,参考:https://www.jieandze1314.com/post/cnposts/28/


ORA实现和优缺点
(2) 常用方法和工具. 目前有许多工具及数据库提供ORA的使用, 包括DAVID, GOstat, Gen- MAPP等. 其中DAVID提供的基因功能集数据库最为全面, 不仅包含大量不同物种的基因功能注释信息, 也涵盖了主流的生物通路注释库如GO条目和KEGG通路, 而且还提供了基因名称转换功能, 及良好的结果展示界面. 因而, DAVID已成为目前应用最广泛的ORA分析工具。
(3) 优缺点. ORA方法基于完备的统计学理论, 具有结果稳健、可靠的优点. 但目前常用的基于统计检验的ORA方法也有一定的局限性, 包括:
(ⅰ) 在对基因进行计数时, 丢失了基因的表达水平或表达差异值等基因属性信息;
(ⅱ) 把通路中的所有基因进行同等对待, 忽视了基因在通路内部生物学意义的不同(如调控和被调控基因的不同)及基因间复杂的相互作用;
(ⅲ) 在获得感兴趣的基因时, 往往需要选取合适的阈值, 而这样有可能会丢失显著性较低但比较关键的基因, 导致检测灵敏性的降低. 为此, 人们需要开发新的富集分析方法来解决这些局限性。
(3)FCS功能集打分:以GSEA基因集富集分析为例
算法原理
相比于针对一组感兴趣的基因通过计数来进行富集分析的ORA方法, 第二代功能富集分析方法FCS的输入数据不仅是全基因组基因, 并且还考虑到每个基因的表达水平或表达差异值等基因属性信息。此外, ORA的检验对象是感兴趣的基因列表与待测基因功能集的共同基因, 而FCS的检验对象则是待测基因功能集中的所有基因。
FCS方法的基本步骤包括:
首先根据案例和对照状态下的基因表达谱对基因组中所有基因表达水平的差异值进行打分或排序, 或直接输入排序好的基因表达谱;
其次是把待测基因功能集中的每个基因的分数通过特定的统计模型转换为待测基因功能集的分数或统计值;
最后利用随机抽样获得的待测基因功能集统计值的背景分布来检验实际观测的统计值的显著水平, 并判断待测基因功能集在案例和对照实验状态下是否发生了统计上的显著变化。
FCS实现和优缺点


总结
这篇推文依旧是一年前的,最近一直在整理仓库文档。
参考前面引用文献,另外补充了点自己知道并收集的,
常用基因功能富集分析方法