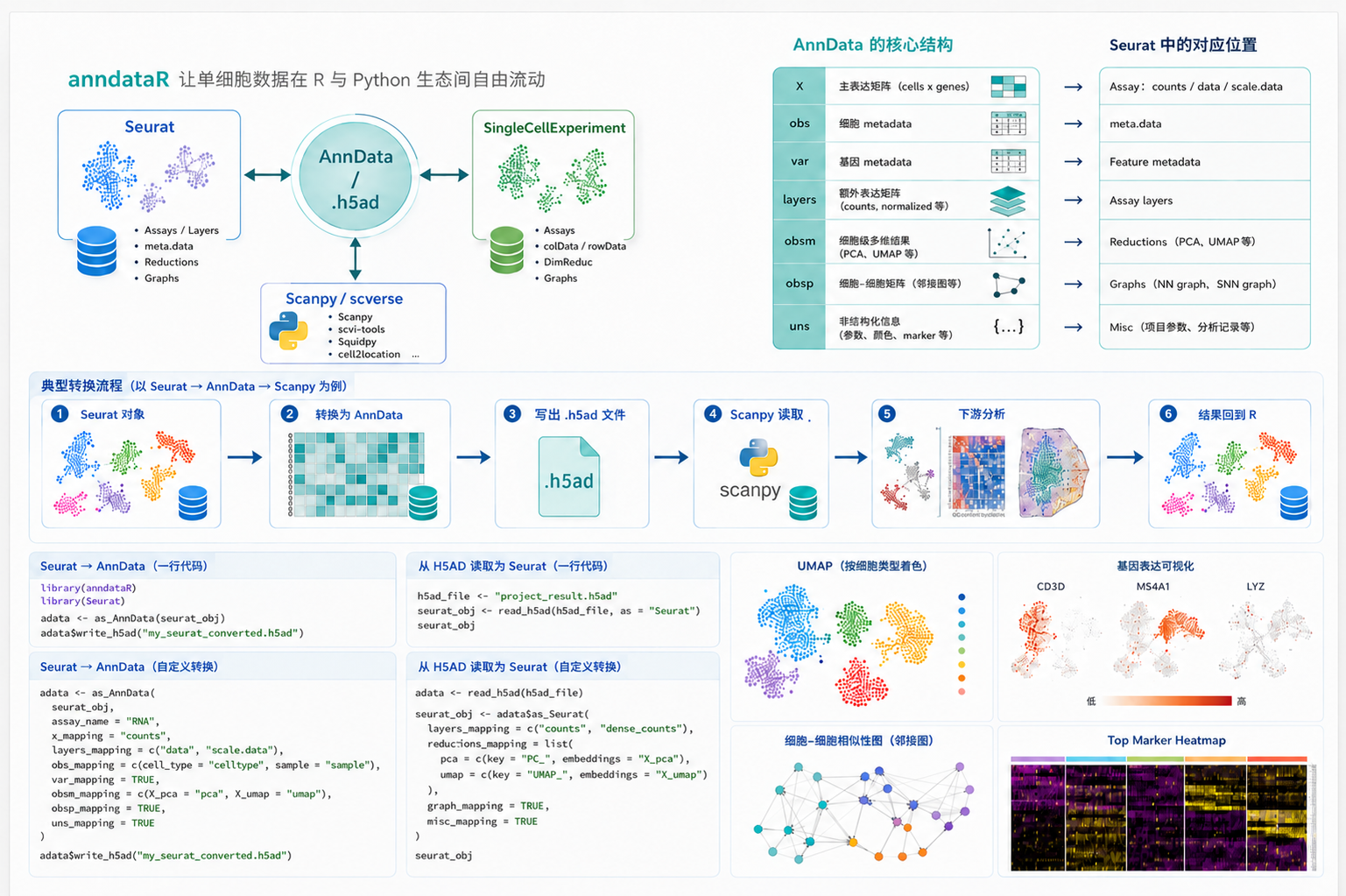
在单细胞组学分析中,数据对象格式往往决定了后续分析流程的可用工具链。R 生态中,Seurat 是应用最广泛的单细胞分析框架之一;Bioconductor 体系中,SingleCellExperiment 是大量方法包采用的标准数据结构;而在 Python 生态中,AnnData 及其 .h5ad 文件格式则是 scanpy、scvi-tools 以及 scverse 生态的核心数据载体。
对于单细胞研究者而言,跨生态协作已经非常常见:上游数据可能来自 Python 流程,下游可视化和注释可能在 R 中完成;或者团队内部使用 Seurat 完成质控、整合、聚类和注释,但投稿、共享或与其他团队协作时又需要交付 .h5ad 文件。过去,这类转换并不总是顺滑。表达矩阵、细胞元数据、基因元数据、降维结果、图结构和额外分析结果都可能在转换过程中丢失或错位。
anndataR 的出现,正是为了解决这一类问题。它为 R 用户提供了原生处理 AnnData 对象的能力,并支持在 AnnData、Seurat 和 SingleCellExperiment 之间进行转换,使 R 生态与 Python scverse 生态之间的数据流转更加规范、透明和可控。
本文将围绕 anndataR 的实际使用场景,系统介绍如何完成 Seurat、AnnData 和 SingleCellExperiment 之间的常见转换,并给出推荐的代码模板与检查方法。
1. 为什么需要 anndataR?
单细胞数据对象并不只是一个表达矩阵。一个完整的分析对象通常包含多个层级的信息:
-
表达矩阵,例如 raw counts、normalized data、scaled data;
-
细胞层面的 metadata,例如样本来源、批次、细胞类型、聚类标签;
-
基因层面的 metadata,例如基因 ID、基因名称、高变基因标记;
-
降维结果,例如 PCA、UMAP、t-SNE;
-
图结构,例如 neighbor graph、shared nearest neighbor graph;
-
其他分析结果,例如 marker gene、参数记录、自定义对象等。
不同生态中的对象结构并不完全一致。以 Seurat 和 AnnData 为例,二者都能表示单细胞数据,但内部字段组织方式不同。Seurat 中常见的 Assay、meta.data、reductions、graphs 和 misc,在 AnnData 中通常对应 X、layers、obs、var、obsm、obsp 和 uns 等字段。
因此,格式转换并不是简单地"另存为一个文件"。一个可靠的转换工具需要明确处理字段映射、矩阵方向、稀疏矩阵格式和对象属性保留等问题。anndataR 的核心价值就在于:它提供了标准化的转换接口,同时允许用户根据具体分析需求自定义映射规则。
2. 三种常见数据结构的关系
在实际项目中,可以将三种对象理解为不同分析生态的核心数据容器:
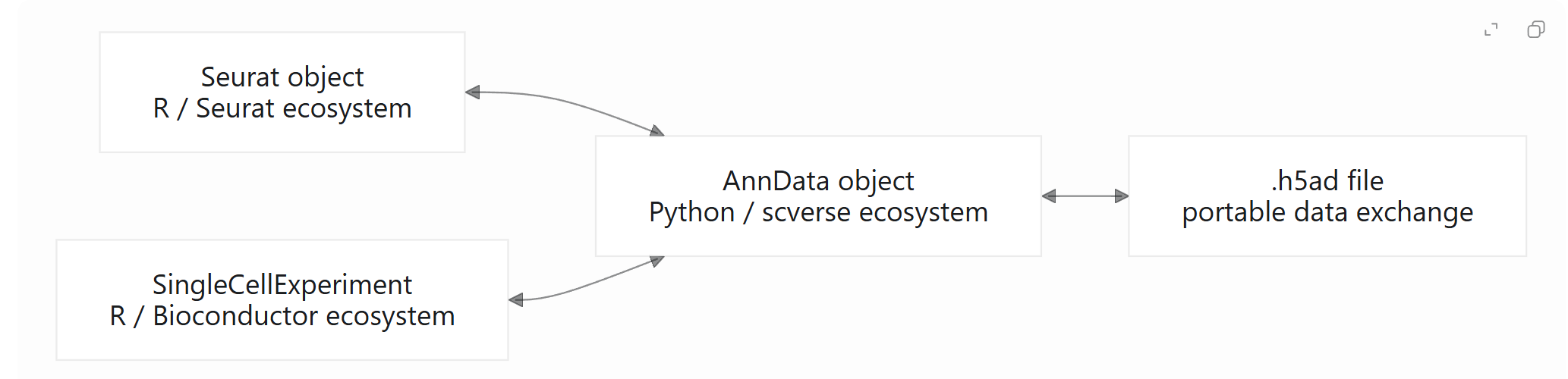
在项目协作中,常见数据流包括:
-
从
Seurat对象导出.h5ad,供 Python 用户使用; -
读取公共数据库发布的
.h5ad文件,并转换为Seurat对象继续分析; -
在
SingleCellExperiment与AnnData之间转换,以便对接 Bioconductor 与 scverse 工具; -
将已有分析对象转换为通用格式,便于数据归档、共享和复现。
3. 安装与加载
anndataR 可以通过 Bioconductor 安装:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("anndataR")如果需要读取或写出 HDF5 格式的 .h5ad 文件,建议同时安装相关依赖:
BiocManager::install("rhdf5")如果需要进行 Seurat 对象转换:
install.packages("Seurat")如果需要使用 SingleCellExperiment:
BiocManager::install("SingleCellExperiment")加载常用包:
library(anndataR)
library(Seurat)4. Seurat 转 AnnData:最简工作流
假设已经在 R 中完成了标准的 Seurat 分析,并得到对象 seurat_obj:
seurat_obj使用 anndataR 转换为 AnnData 对象非常直接:
adata <- as_AnnData(seurat_obj)
adata随后可以写出为 .h5ad 文件:
adata$write_h5ad("my_seurat_converted.h5ad")在 Python 中,可以使用 scanpy 读取:
import scanpy as sc
adata = sc.read_h5ad("my_seurat_converted.h5ad")
adata对于常规场景,以上代码已经能够覆盖大多数需求。尤其是在需要将 Seurat 分析结果交付给 Python 用户时,这种方式非常简洁。
5. 自定义 Seurat 到 AnnData 的转换过程
在更复杂的分析项目中,用户往往希望明确指定哪些内容进入 AnnData 的不同字段。例如,希望将 counts 写入 X,将部分矩阵写入 layers,将 PCA 和 UMAP 写入 obsm,并保留部分 metadata。
示例代码如下:
adata <- as_AnnData(
seurat_obj,
assay_name = "RNA",
x_mapping = "counts",
layers_mapping = c("dense_counts"),
obs_mapping = c(
RNA_count = "nCount_RNA",
metadata1 = "metadata1"
),
var_mapping = FALSE,
obsm_mapping = list(
X_pca = "pca",
X_umap = "umap"
),
obsp_mapping = TRUE,
uns_mapping = c("misc1", "misc2")
)
adata写出结果:
adata$write_h5ad("./my_seurat_converted.h5ad")上述参数的作用可以概括为:
-
assay_name:指定需要转换的 Seurat assay; -
x_mapping:指定 AnnData 中X的来源; -
layers_mapping:指定需要写入 AnnDatalayers的矩阵; -
obs_mapping:指定细胞 metadata 的映射; -
var_mapping:控制基因 metadata 的转换; -
obsm_mapping:指定降维结果的映射,例如 PCA、UMAP; -
obsp_mapping:控制图结构的映射; -
uns_mapping:指定非结构化信息的映射。
这种显式映射方式更适合正式项目、数据交付和可复现分析。它的优势是可读性更高,也更容易在后续检查时定位问题。
需要注意的是,写出 .h5ad 文件时通常只需要调用一次 write_h5ad():
adata$write_h5ad("./my_seurat_converted.h5ad")不建议写成嵌套调用:
adata$write_h5ad(adata$write_h5ad("./my_seurat_converted.h5ad"))后者会重复执行写出操作,并不符合常规用法。
6. Seurat 与 AnnData 的字段映射关系
理解字段映射有助于减少转换过程中的不确定性。下面是一个简化的结构关系:
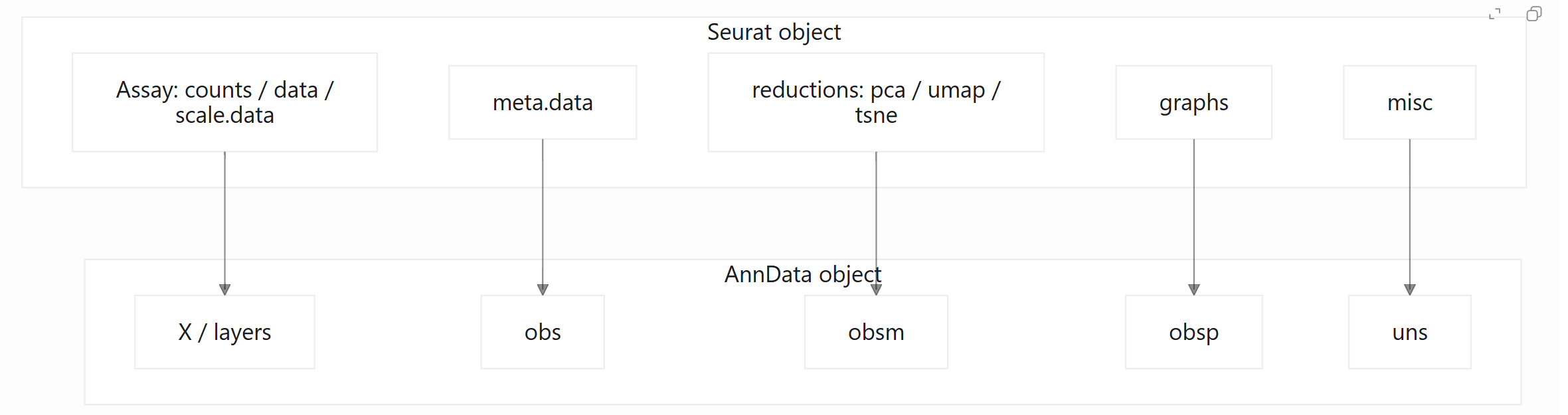
其中,obs 通常存储细胞层面的信息,var 通常存储基因层面的信息,obsm 通常用于保存 PCA、UMAP 等细胞嵌入结果,obsp 常用于保存细胞之间的图或距离矩阵,uns 则用于保存非结构化信息。
在正式转换前,建议先明确三个问题:
-
哪个表达矩阵应该作为
AnnData.X? -
哪些矩阵应该进入
AnnData.layers? -
降维、图结构和元数据是否需要完整保留?
对于下游以差异分析或重新归一化为主的场景,通常应优先保留 raw counts。对于下游以可视化和细胞类型注释复核为主的场景,则应确保 UMAP、cluster 和细胞注释等信息完整写出。
7. 读取 H5AD 文件为 Seurat 对象
除了从 Seurat 导出 .h5ad,另一个常见需求是读取外部 .h5ad 文件,并转换为 Seurat 对象。
可以使用官方示例文件:
h5ad_file <- system.file(
"extdata",
"example.h5ad",
package = "anndataR"
)一行代码读取为 Seurat:
seurat_obj <- read_h5ad(h5ad_file, as = "Seurat")
seurat_obj也可以先读取为 AnnData,再转换为 Seurat:
adata <- read_h5ad(h5ad_file)
seurat_obj <- adata$as_Seurat()
seurat_obj如果需要更精细地控制转换过程,可以使用自定义映射:
seurat_obj <- adata$as_Seurat(
layers_mapping = c("counts", "dense_counts"),
object_metadata_mapping = c(
metadata1 = "Int",
metadata2 = "Float"
),
assay_metadata_mapping = FALSE,
reduction_mapping = list(
pca = c(
key = "PC_",
embeddings = "X_pca",
loadings = "PCs"
),
umap = c(
key = "UMAP_",
embeddings = "X_umap"
)
),
graph_mapping = TRUE,
misc_mapping = c(
misc1 = "Bool",
misc2 = "IntScalar"
)
)
seurat_obj这类自定义映射尤其适合公共数据再分析。因为不同数据发布者对 .h5ad 字段命名习惯并不完全一致,例如 UMAP 可能存储为 X_umap,PCA 可能存储为 X_pca,不同 layer 也可能采用不同命名。显式指定映射规则能够降低误读字段的风险。
8. SingleCellExperiment 与 AnnData 的转换
Bioconductor 生态中的 SingleCellExperiment 同样是单细胞分析的重要数据结构。anndataR 也支持它与 AnnData 之间的转换。
读取 .h5ad 为 SingleCellExperiment:
library(anndataR)
library(SingleCellExperiment)
h5ad_file <- system.file(
"extdata",
"example.h5ad",
package = "anndataR"
)
sce_obj <- read_h5ad(h5ad_file, as = "SingleCellExperiment")
sce_obj或者先读取为 AnnData:
adata <- read_h5ad(h5ad_file)
sce_obj <- adata$as_SingleCellExperiment()
sce_obj将 SingleCellExperiment 转为 AnnData:
adata <- as_AnnData(sce_obj)
adata$write_h5ad("my_sce_converted.h5ad")也可以直接写出:
write_h5ad(sce_obj, "my_sce_converted.h5ad")对于以 Bioconductor 方法包为主的分析流程,这一功能非常实用。例如,用户可以在 SingleCellExperiment 中完成特定统计分析,再将结果导出为 .h5ad,供 Python 生态继续处理。
9. 转换后的质量检查
格式转换完成并不意味着流程结束。为了保证数据在跨生态流转后仍然可靠,建议至少进行以下检查。
9.1 检查维度
dim(seurat_obj)
adata$shape需要确认细胞数和基因数是否符合预期。由于不同对象对矩阵方向的表示方式可能不同,检查维度时应特别留意行列含义。
9.2 检查细胞元数据
head(seurat_obj@meta.data)
head(adata$obs)重点检查样本信息、批次信息、细胞类型注释和聚类标签是否完整。
9.3 检查降维结果
Reductions(seurat_obj)
adata$obsm_keys()如果下游需要复用 UMAP 或 PCA,应确认对应字段已经正确保留。
9.4 绘制 UMAP 进行直观确认
DimPlot(
seurat_obj,
reduction = "umap",
group.by = "seurat_clusters"
)也可以手动提取嵌入结果并使用 ggplot2 绘图:
library(ggplot2)
umap_df <- as.data.frame(Embeddings(seurat_obj, "umap"))
umap_df$cluster <- seurat_obj$seurat_clusters
ggplot(umap_df, aes(UMAP_1, UMAP_2, color = cluster)) +
geom_point(size = 0.6, alpha = 0.8) +
theme_classic() +
labs(
title = "UMAP after H5AD to Seurat conversion",
x = "UMAP 1",
y = "UMAP 2"
)图 3. 转换后通过 UMAP 检查细胞结构和聚类标签是否保留
10. 推荐实践
在实际项目中,建议根据数据复杂度选择不同的转换策略。
对于结构简单、只需要完成基本交付的数据,可以优先使用默认接口:
adata <- as_AnnData(seurat_obj)
adata$write_h5ad("converted.h5ad")或者:
seurat_obj <- read_h5ad("dataset.h5ad", as = "Seurat")对于正式项目、跨团队协作或公共数据发布,更推荐使用显式 mapping。这样可以清楚记录每个字段的来源和去向,也便于后续复现。
此外,建议保留转换脚本,而不是只保留转换后的文件。转换脚本相当于数据结构说明文档,能够帮助后续使用者理解 .h5ad 文件中的 X、layers、obs、obsm 等字段具体来自哪里。
11. 小结
Seurat、AnnData 和 SingleCellExperiment 分别服务于不同的单细胞分析生态。随着跨平台协作和公共数据复用越来越普遍,可靠的数据对象转换已经成为单细胞分析流程中的基础环节。
anndataR 为 R 用户提供了一个清晰、规范的解决方案。它既能用简洁接口完成常规转换,也能通过自定义映射满足复杂项目需求。对于需要在 R 与 Python 之间传递单细胞数据的研究者而言,anndataR 可以显著减少对象转换中的不确定性,提高数据共享和分析复现的效率。
以后,当需要将 Seurat 对象交付给 Python 用户时,可以从下面这段代码开始:
library(anndataR)
adata <- as_AnnData(seurat_obj)
adata$write_h5ad("converted_from_seurat.h5ad")当需要把 .h5ad 文件接入 Seurat 分析流程时,可以使用:
library(anndataR)
library(Seurat)
seurat_obj <- read_h5ad("dataset.h5ad", as = "Seurat")如果项目涉及更复杂的数据结构,则建议进一步使用 as_AnnData() 或 as_Seurat() 中的 mapping 参数,明确控制表达矩阵、元数据、降维结果和图结构的转换方式。
参考资料
-
anndataR 官方文档:https://anndatar.scverse.org/
-
Read/write Seurat objects using anndataR:https://anndatar.scverse.org/articles/usage_seurat.html
-
Read/write SingleCellExperiment objects using anndataR:https://anndatar.scverse.org/articles/usage_singlecellexperiment.html