Cell Ranger
Cell Ranger是用于10x单细胞转录组数据处理一套Linux工具集,包含数据比对,生成表达矩阵,聚类分析和图形可视化等多个功能。一般用cell ranger进行上游分析。
官网:https://www.10xgenomics.com/support/software/cell-ranger/latest
由于测序仪器的测序能力远大于测试样本序列量,为避免仪器浪费,因此一个lane同时测定多个样品成为很自然的思路。然而为了区分多种样品的序列,就必须要给不同样品加上特定的"标签",从而可以在后续数据分析时将不同样品数据分开,而这个"标签"就是barcode。10xBarcode是一段16nt的核苷酸序列(序列空间350万),在每一个Gel Beads中的Barcode序列都是一致的,在后面Barcode与细胞融合形成水凝珠之后,可以保证一个细胞的所有基因序列都带着相同的Barcode序列,也就可以认定这些序列来自同一个细胞。所以我们通常说Barcode序列是用来标记细胞的。Cellranger主要就是用于区分barcode的识别与所连reads的定量。
UMI(unique molecular identifier分子标签)
UMI是一段12nt的核苷酸序列(序列空间100万),但与Barcode序列不同的是,一个Gel Beads中UMI序列是不同的。UMI序列的空间很大,远多于需要检测的原始细胞的mRNA数量,(即使一种mRNA有多条,也是达不到UMI的序列空间的)。所以每一条mRNA都会带上一个独特的UMI。UMI的最大作用是去重和绝对定量。
可以这样理解:barcode是每个凝胶微珠的身份证号码;UMI是每个DNA标签分子的身份证号码
软件下载
mkdir cellranger #在目标路径下新建文件夹
cd cellranger
#下载软件
wget -O cellranger-8.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.0.tar.gz?Expires=1714073771&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=CHmp~VHwcV2qCWGIk-wiR-LIT2FVFVny09DXCrksxPRGxI8llh6N87Z2kd4RQin5TH57AlpIjfh5iMdDw54pB3u7oMzANtgdQLd1AUA8FlPzikAvW6Kv02yCwvlpiGnFUwzYnr3aJuATgdgOJLT6RGJumRGt2PQVim45u1jkJ~DeahmRIuntllk8QJ4sOIHqAPvYoPTQ47NN5HXlqMIbw1K8-W7SHMTIXJ4PDudwblqf6xCJltxcLob1P2vD9nwutSsJrdvyaEblv1ZjPGFg5fXkw0Yk8H0He4MRGdhxDTLRgP2~Svneje4yglVQCu~Xe5Yd-UybpW6mhHiTx0GFdg__"
#解压
tar -zxvf cellranger-8.0.0.tar.gz
#添加环境量
export PATH=./path/cellranger/cellranger-8.0.0:$PATH
#进入cellranger
cellranger成功后返回如下:
常用命令-mkgtf&mkref 建立索引文件
GTF文件(共9列):是对基因组进行注释的
mkgtf:Raw gtf---mkgtf---filtered gtf,从网上下载的GTF文件几乎包含所有基因,可以利用此命令将不需要的信息过滤掉。
常用命令-mkfastq 格式转换
可以用cellranger mkfastq将BCL格式文件转换为fastq文件。
BCL:测序仪得到的初始格式(测序公司才用的到,普通用户用不到)。
注意:在定量之前,必须先将格式按照要求修改,cell range对格式要求严格,正确格式如下:
[Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz
Read Type:
T1:Sample index read(optional)
T2:Sample index read(optional)
R1:Read1
R2:Read2
eg:
Test_S1_L001_R1_001.fastq.gz
Test_S1_L001_R2_001.fastq.gz常用命令-count:对原始数据进行比对定量,最后得到基因-细胞的表达矩阵
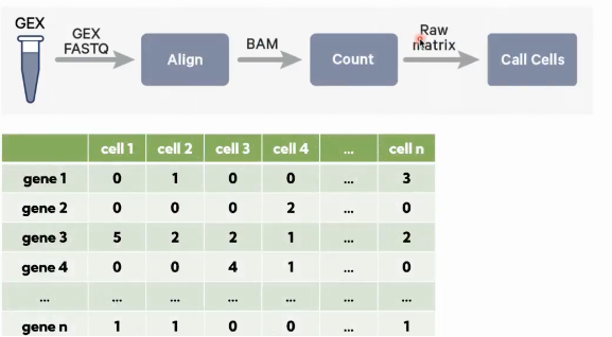
语法:
cellranger count--id #输出目录名
--transcriptome #基因组索引文件路径
--fastqs #FASTQ数据存放路径
--sample #需要运行的样本名称
--include-introns #定量时是否包含内含子(7.0版本默认为True)
#下面非必选项
--lanes #指定lane编号
--no-bam #不输出Bam文件
--nosecondary #不进行下游分析(仅定量)
--ocalcores #指定最大核心数
--localmem #指定最大内存(GB)例如:
cellranger count--id sample_test
--transcriptome /home/wangyan/cellranger/refdata-gex-GRCh38-2020-A
--fastqs /home/wangyan/cellranger/sample_fastqs
--sample Sample_1
--include-introns false如果运行成功会出现以下这串代码:
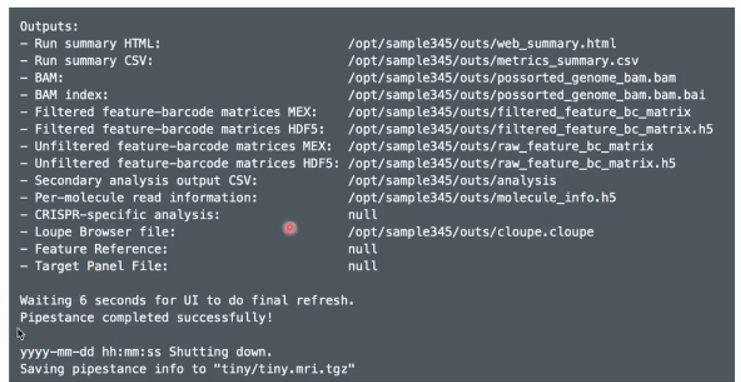
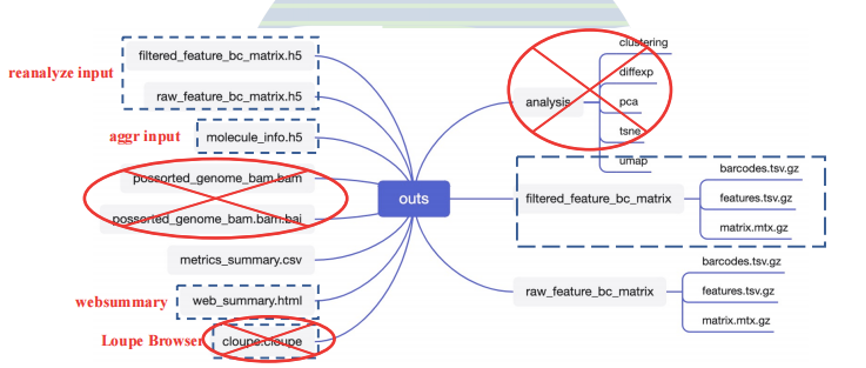
输出的文件包括以下内容:
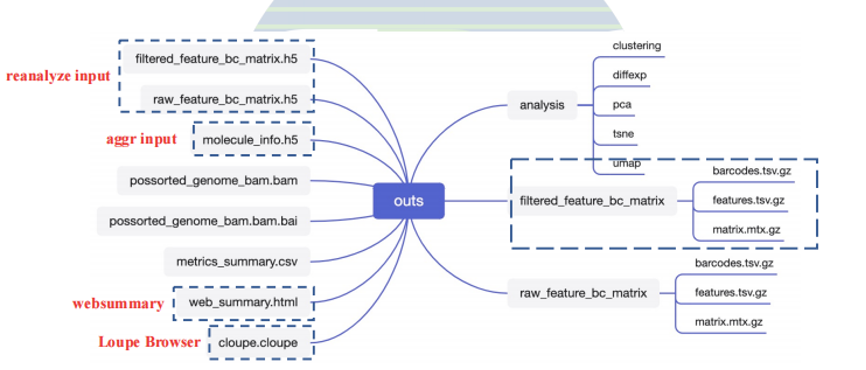
其中的reanalyze input文件可以用于下游的Seurat分析。
如果在前面的命令中设置了--nosecondary fasle,则cellranger不进行进一步下游分析: