接着上次的内容,上篇内容给大家分享了基因表达量怎么做分组差异分析,从而获得差异基因集,想了解的可以去看一下,这篇主要给大家分享一下得到显著差异基因集后怎么做一下通路富集。
1.准备差异基因集
我就直接把上次分享的拿到这边了。我们一般都把差异基因分为上调基因和下调基因分别做通路富集分析。下面上代码,可能包含我的一些个人习惯,勿怪。显著差异基因的筛选条件根据个人需求设置哈。
##载入所需R包
library(readxl)
library(DOSE)
library(org.Hs.eg.db)
library(topGO)
library(pathview)
library(ggplot2)
library(GSEABase)
library(limma)
library(clusterProfiler)
library(enrichplot)
##edger
edger_diff <- diff_gene_Group
edger_diff_up <- rownames(edger_diff[which(edger_diff$logFC > 0.584962501),])
edger_diff_down <- rownames(edger_diff[which(edger_diff$logFC < -0.584962501),])
##deseq2
deseq2_diff <- diff_gene_Group2
deseq2_diff_up <- rownames(deseq2_diff[which(deseq2_diff$log2FoldChange > 0.584962501),])
deseq2_diff_down <- rownames(deseq2_diff[which(deseq2_diff$log2FoldChange < -0.584962501),])
##将差异基因集保存为一个list
gene_diff_edger_deseq2 <- list()
gene_diff_edger_deseq2[["edger_diff_up"]] <- edger_diff_up
gene_diff_edger_deseq2[["edger_diff_down"]] <- edger_diff_down
gene_diff_edger_deseq2[["deseq2_diff_up"]] <- deseq2_diff_up
gene_diff_edger_deseq2[["deseq2_diff_down"]] <- deseq2_diff_down2.进行通路富集分析
这里主要介绍普通的GO&KEGG&GSEA的简单富集。筛选显著富集通路的筛选条件也是根据自己的需求决定,一般是矫正后P值小于0.05。我这里是省事,写了各list循环。
for (i in 1:length(gene_diff_edger_deseq2)){
keytypes(org.Hs.eg.db)
entrezid_all = mapIds(x = org.Hs.eg.db,
keys = gene_diff_edger_deseq2[[i]],
keytype = "SYMBOL", #输入数据的类型
column = "ENTREZID")#输出数据的类型
entrezid_all = na.omit(entrezid_all) #na省略entrezid_all中不是一一对应的数据情况
entrezid_all = data.frame(entrezid_all)
##GO富集##
GO_enrich = enrichGO(gene = entrezid_all[,1],
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID", #输入数据的类型
ont = "ALL", #可以输入CC/MF/BP/ALL
#universe = 背景数据集我没用到它。
pvalueCutoff = 1,qvalueCutoff = 1, #表示筛选的阈值,阈值设置太严格可导致筛选不到基因。可指定 1 以输出全部
readable = T) #是否将基因ID映射到基因名称。
GO_enrich_data = data.frame(GO_enrich)
write.csv(GO_enrich_data,paste('GO_enrich_',names(gene_diff_edger_deseq2)[i], '.csv', sep = ""))
GO_enrich_data <- GO_enrich_data[which(GO_enrich_data$p.adjust < 0.05),]
write.csv(GO_enrich_data,paste('GO_enrich_',names(gene_diff_edger_deseq2)[i], '_filter.csv', sep = ""))
###KEGG富集分析###
KEGG_enrich = enrichKEGG(gene = entrezid_all[,1], #即待富集的基因列表
keyType = "kegg",
pAdjustMethod = 'fdr', #指定p值校正方法
organism= "human", #hsa,可根据你自己要研究的物种更改,可在https://www.kegg.jp/brite/br08611中寻找
qvalueCutoff = 1, #指定 p 值阈值(可指定 1 以输出全部)
pvalueCutoff=1) #指定 q 值阈值(可指定 1 以输出全部)
KEGG_enrich_data = data.frame(KEGG_enrich)
write.csv(KEGG_enrich_data, paste('KEGG_enrich_',names(gene_diff_edger_deseq2)[i], '.csv', sep = ""))
KEGG_enrich_data <- KEGG_enrich_data[which(KEGG_enrich_data$p.adjust < 0.05),]
write.csv(KEGG_enrich_data, paste('KEGG_enrich_',names(gene_diff_edger_deseq2)[i], '_filter.csv', sep = ""))
}3.通路富集情况可视化
这里只介绍一种简单的气泡图,当然还有其他的自己去了解吧。
##GO&KEGG富集BPCCMFKEGG分面绘图需要分开处理一下,富集结果里的ONTOLOGYL列修改
GO_enrich_data_BP <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "BP")
GO_enrich_data_CC <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "CC")
GO_enrich_data_MF <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "MF")
##提取GO富集BPCCMF的top5
GO_enrich_data_filter <- rbind(GO_enrich_data_BP[1:5,], GO_enrich_data_CC[1:5,], GO_enrich_data_MF[1:5,])
##重新整合进富集结果
GO_enrich@result <- GO_enrich_data_filter
##处理KEGG富集结果
KEGG_enrich@result <- KEGG_enrich_data
ncol(KEGG_enrich@result)
KEGG_enrich@result$ONTOLOGY <- "KEGG"
KEGG_enrich@result <- KEGG_enrich@result[,c(10,1:9)]
##整合GO KEGG富集结果
ego_GO_KEGG <- GO_enrich
ego_GO_KEGG@result <- rbind(ego_GO_KEGG@result, KEGG_enrich@result[1:5,])
ego_GO_KEGG@result$ONTOLOGY <- factor(ego_GO_KEGG@result$ONTOLOGY, levels = c("BP", "CC", "MF","KEGG"))##规定分组顺序
##简单画图
pdf("edger_diff_up_dotplot.pdf", width = 7, height = 7)
dotplot(ego_GO_KEGG, split = "ONTOLOGY", title="UP-GO&KEGG", label_format = 60, color = "pvalue") +
facet_grid(ONTOLOGY~., scale = "free_y")+
theme(plot.title = element_text(hjust = 0.5, size = 13, face = "bold"), axis.text.x = element_text(angle = 90, hjust = 1))
dev.off()4.气泡图如图所示

做了些处理,真实图片,左侧pathway是跟后面气泡一一对应的,当然还有其他可视化方式那就需要各位自己去探索了,谢谢!
5.GSEA富集分析
这里也是做一下简单的GSEA
##GSEA官方网站下载背景gmt文件并读入
geneset <- list()
geneset[["c2_cp"]] <- read.gmt("c2.cp.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_kegg_legacy"]] <- read.gmt("c2.cp.kegg_legacy.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_kegg_medicus"]] <- read.gmt("c2.cp.kegg_medicus.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_reactome"]] <- read.gmt("c2.cp.reactome.v2023.2.Hs.symbols.gmt")
geneset[["c3_tft"]] <- read.gmt("c3.tft.v2023.2.Hs.symbols.gmt")
geneset[["c4_cm"]] <- read.gmt("c4.cm.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_bp"]] <- read.gmt("c5.go.bp.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_cc"]] <- read.gmt("c5.go.cc.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_mf"]] <- read.gmt("c5.go.mf.v2023.2.Hs.symbols.gmt")
geneset[["c6"]] <- read.gmt("c6.all.v2023.2.Hs.symbols.gmt")
geneset[["c7"]] <- read.gmt("c7.all.v2023.2.Hs.symbols.gmt")
##进行GSEA富集分析,这里也是写了个循环
gsea_results <- list()
for (i in names(gene_diff)){
geneList <- gene_diff[[i]]$logFC
names(geneList) <- toupper(rownames(gene_diff[[i]]))
geneList <- sort(geneList,decreasing = T)
for (j in names(geneset)){
listnames <- paste(i,j,sep = "_")
gsea_results[[listnames]] <- GSEA(geneList = geneList,
TERM2GENE = geneset[[j]],
verbose = F,
pvalueCutoff = 0.1,
pAdjustMethod = "none",
eps=0)
}
}
##批量绘图,注意这里如果有空富集通路,会报错!
for (j in 1:nrow(gsea_results[[i]]@result)) {
p <- gseaplot2(x=gsea_results[[i]],geneSetID=gsea_results[[i]]@result$ID[j], title =
gsea_results[[i]]@result$ID[j])
pdf(paste(paste(names(gsea_results)[i], gsea_results[[i]]@result$ID[j], sep =
"_"),".pdf",sep = ""))
print(p)
dev.off()
}6.GSEA富集最简单图形如下
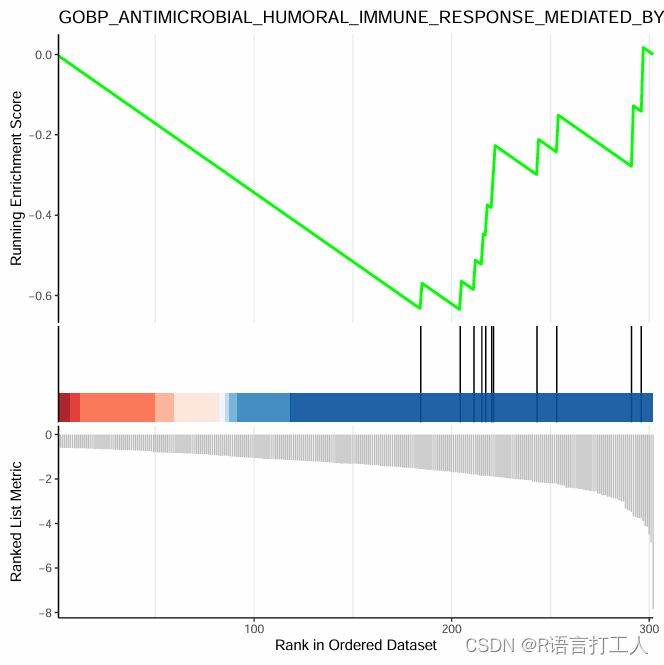
分享到此结束了,希望对大家有所帮助。