
引言
本系列开启R中单细胞RNA-seq数据分析教程[1],持续更新,欢迎关注,转发!
10. 伪时间细胞排序
如前所述,在 UMAP 嵌入中看到的背侧端脑细胞形成的类似轨迹的结构,很可能代表了背侧端脑兴奋性神经元的分化和成熟过程。这个过程很可能是连续的,因此将其视为一个连续的轨迹,而非不同的聚类,更为合适。在这种情况下,进行所谓的伪时间细胞排序或伪时间分析会更加具有信息价值。
目前,有许多不同的伪时间分析方法。常见的包括扩散图(在 R 的 destiny 包中实现)和 monocle。在这里,将展示如何使用 destiny 对数据中的背侧端脑细胞进行伪时间分析。
首先,提取感兴趣的细胞。接下来,会重新识别这些细胞的高变基因,因为在这些细胞之间,代表背侧端脑细胞与其他细胞的差异的基因不再是有用的。
seurat_dorsal <- subset(seurat, subset = RNA_snn_res.1 %in% c(0,2,5,6,10))
seurat_dorsal <- FindVariableFeatures(seurat_dorsal, nfeatures = 2000)正如你可能已经注意到的,背侧端脑神经前体细胞(NPC)中有两个聚类与第三个聚类分开,原因是它们处于不同的细胞周期阶段。由于关注的是分化和成熟过程中的整体分子变化,细胞周期的变化可能会对分析产生较大的干扰。因此,可以尝试通过排除与细胞周期相关的基因,来减少细胞周期对分析结果的影响。
VariableFeatures(seurat) <- setdiff(VariableFeatures(seurat), unlist(cc.genes))接下来,可以创建一个新的 UMAP 嵌入,并绘制一些特征图来检查数据的表现。
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
结果不是很理想。G2M 细胞不再集中在单独的聚类中,但它仍然干扰了细胞类型分化的轨迹。例如,EOMES+ 细胞被分布在两个不同的群体中。需要进一步减少细胞周期的影响。
如上所述,ScaleData 函数提供了一个选项,可以将表示不必要变异来源的变量纳入其中。可以尝试利用这个选项,进一步减少细胞周期的影响;不过在这之前,需要为每个细胞生成细胞周期相关的评分,以描述它们的细胞周期状态。
seurat_dorsal <- CellCycleScoring(seurat_dorsal,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes,
set.ident = TRUE)
seurat_dorsal <- ScaleData(seurat_dorsal, vars.to.regress = c("S.Score", "G2M.Score"))接下来,可以创建一个新的 UMAP 嵌入,并绘制一些特征图来检查数据的表现。
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)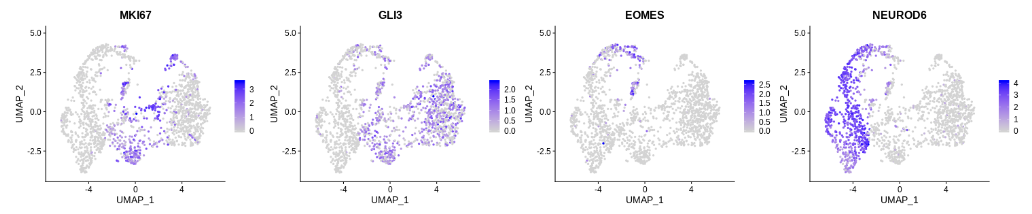
虽然结果不是完全理想,但至少不再看到两个分开的 EOMES+ 细胞群体。
接下来,让尝试运行扩散图,以便对细胞进行排序。
library(destiny)
dm <- DiffusionMap(Embeddings(seurat_dorsal, "pca")[,1:20])
dpt <- DPT(dm)
seurat_dorsal$dpt <- rank(dpt$dpt)
FeaturePlot(seurat_dorsal, c("dpt","GLI3","EOMES","NEUROD6"), ncol=4)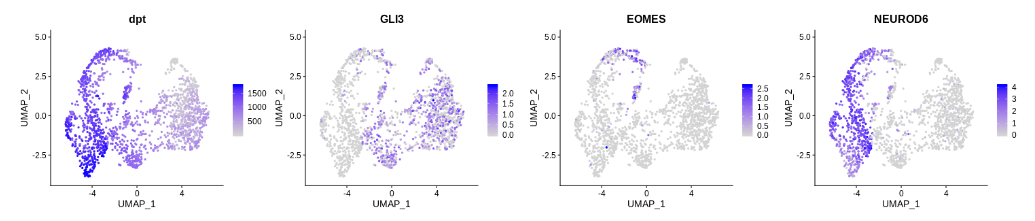
为了展示沿着构建的伪时间的表达变化,带有拟合曲线的散点图通常是一种直观的方法。
if (is(seurat_dorsal[['RNA']], 'Assay5')){
expr <- LayerData(seurat_dorsal, assay = "RNA", layer = "data")
} else{
expr <- seurat_dorsal[['RNA']]@data
}
library(ggplot2)
plot1 <- qplot(seurat_dorsal$dpt, as.numeric(expr["GLI3",]),
xlab="Dpt", ylab="Expression", main="GLI3") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot2 <- qplot(seurat_dorsal$dpt, as.numeric(expr["EOMES",]),
xlab="Dpt", ylab="Expression", main="EOMES") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot3 <- qplot(seurat_dorsal$dpt, as.numeric(expr["NEUROD6",]),
xlab="Dpt", ylab="Expression", main="NEUROD6") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot1 + plot2 + plot3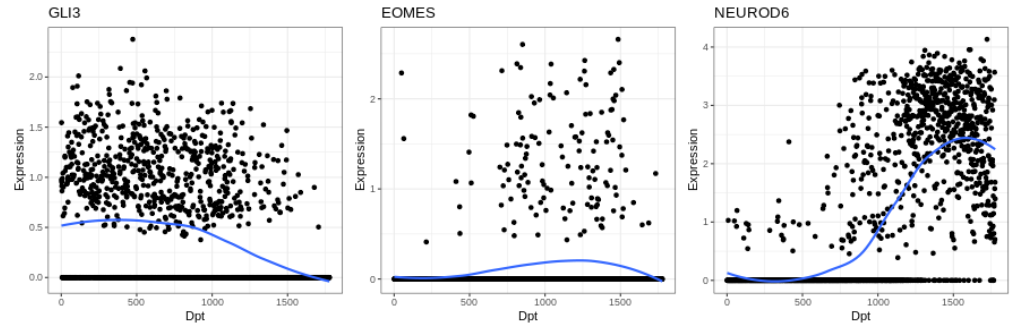
11. 结果保存
这就是本教程第一部分的全部内容,涵盖了对单个 scRNA-seq 数据集进行的大部分基础分析。在分析结束时,当然希望保存结果,可能是操作过一段时间的 Seurat 对象,这样下次就不需要重新运行所有分析了。保存 Seurat 对象的方法与保存其他 R 对象一样。可以使用 saveRDS/readRDS 来分别保存和加载 Seurat 对象。
saveRDS(seurat, file="DS1/seurat_obj_all.rds")
saveRDS(seurat_dorsal, file="DS1/seurat_obj_dorsal.rds")
seurat <- readRDS("DS1/seurat_obj_all.rds")
seurat_dorsal <- readRDS("DS1/seurat_obj_dorsal.rds")或者可以使用 save/load 来一起保存或加载多个对象。
save(seurat, seurat_dorsal, file="DS1/seurat_objs.rdata")
load("DS1/seurat_objs.rdata")未完待续,欢迎关注!
动动您发财的小手点个赞吧!欢迎转发!
Reference 1
Source: https://github.com/quadbio/scRNAseq_analysis_vignette
本文由mdnice多平台发布