原文标题:An ensemble deep learning framework for multi-class LncRNA subcellular localization with innovative encoding strategy
原文链接:
lncRNA亚细胞定位研究方法,主要分为三种类型:基于传统特征的方法、基于深度学习的方法和混合方法。
( i )传统的基于特征的方法:这些方法依赖于序列特征进行预测。
Cao等人引入了lncLocator ,使用4-mer频率特征结合堆叠自编码器、随机森林和SVM。
Su等人提出了iLoc-lncRNA ,该方法使用8-mer频率特征、二项分布进行特征选择和SVMs。
Gudenas和Wang开发了DeepLncRNA,它利用了深度学习、2-5-mer特征、RNA结合基序和基因组位置。
Ahmad等人提出了Locate-R,使用局部深度SVMs,选择了655个最优的k-mer特征。
Fan等人引入了lncLocPred,一种基于逻辑回归的预测器,使用k-mer,PseKNC和Triplet特征。
lncLocation 集成了多个特征和自动编码器进行特征提取和混合选择。
Zhang等人将iLoc-lncRNA改进到2.0版本,增加了基于互信息和增量特征选择策略。
( ii )基于深度学习的方法:这些方法使用深度学习模型,包括CNN和基于图的网络。
Zeng等人开发了DeepLncLoc,基于使用子序列嵌入技术的文本CNN。
Jeon等人引入了TACOS,一种基于树的堆叠分类器,用于预测lncRNA在10种细胞类型中的定位。
Li等人提出了GraphLncLoc,将lncRNA序列转换为图并使用图卷积网络。
Zeng等人还提出了LncLocFormer,使用8个Transformer块对长程序列依赖关系进行建模,并使用了定位特定的注意力机制。
Li等人开发了SGCL-LncLoc,将序列转换为de Bruijn图,使用Word2Vec进行节点表示,并通过图卷积网络进行精炼。
( iii )混合方法:这些方法结合了基于特征和深度学习的方法。
Yuan等人提出了RNALight ,使用k-mer特征和LightGBM进行mRNA和lncRNA定位预测。Yang等引入lncSLPre,综合序列组成、理化性质和结构数据,结合分类器输出。Wang等人微调了一个预训练的多任务RNA结合蛋白模型来开发DeepLocRNA,用于预测各种RNA的亚细胞定位。
现有的计算方法面临两个主要的限制:
( i )它们往往无法捕获位置特异性的核苷酸分布模式,而这对于区分定位信号是必不可少的;
大多数预测器使用k-mer特征编码原始lncRNA序列。单纯使用k-mer数值表示特征无法保留原始lncRNA序列的序列顺序信息,无法捕捉核苷酸的位置特异性分布信息。
( ii )它们缺乏一种有效的特征表示策略,可以跨多个亚细胞区室进行泛化。
为了应对这些挑战,提出了一种新的深度学习框架MGBLncLoc,该框架集成了一种新设计的编码策略------多类核苷酸分布更新广义编码( MCD-ND ) 。与传统的基于k-mer频率的编码方式将序列视为简单的频率分布不同,MCD-ND显式地建模了核苷酸在不同位置的分布密度,从而保留了序列的组成和空间组织 。这种编码机制增强了定位类之间的特征区分性,提高了模型的可解释性和预测精度。为了进一步利用MCD-ND的独特特性,我们设计了一个包含多个专用神经网络模块的深度学习架构。具体来说,模型集成了多Dconv(可变形卷积)头转置注意力( MDTA )用于捕获层次序列依赖 ,门控Dconv前馈网络( GDFN )用于精炼特征表示 ,卷积神经网络( CNNs )用于提取局部序列模体 ,双向门控循环单元( BiGRUs )用于建模长距离上下文依赖。这种协同组合使得MGBLncLoc能够有效地学习全局和局部序列特征。
两个创新点:
(1) MGBLncLoc模型纳入了独特的编码器MCD-ND,能够更精确地表示序列中的核苷酸区域,区分保守区域和歧视性区域。MCD-ND编码器不仅考虑了序列中不同核苷酸组之间的结构特征,而且强调了对整个序列中核苷酸分布的分析。
(2) MGBLncLoc模型集成了多个先进的DNN模块。
方法
数据集
在目前的lncRNA亚细胞定位研究中,研究人员主要构建了三个基准数据集:一个是由Zeng等人从RNALocate 1.0数据库中导出的五个亚细胞区室的数据,另一个是由Yang等人构建的两个亚细胞区室的数据,以及其他研究人员构建的四个亚细胞区室数据集。为了获得可靠的数据集,本研究遵循先前研究的方法,从RNALocate 2.0数据库中下载已知的lncRNA亚细胞定位序列 。RNALocate 2.0数据库包含了超过21万个与RNA相关的亚细胞定位条目和实验证据的记录,涵盖了104个物种的171个亚细胞定位中的超过11万个RNA。与1.0版本相比,2.0版本扩大了数据来源和物种覆盖范围。我们下载了9256条与lncRNA相关的亚细胞定位序列,其中一些属于多个亚细胞定位。因此,仅保留位于单隔室的数据,以客观评估模型的预测能力。序列通常被划分为多个条目,如果它们具有相同的基因符号,则合并这些条目。得到的数据集包含分布在10个亚细胞定位中的lncRNA序列。由于内质网、线粒体、突触、核质和外泌体等特定细胞定位的序列数量不足,不足以构建具有统计学意义的基准数据集,因此排除了这些亚细胞定位的序列。**为了避免冗余和减少同源偏倚,同时保留原始分布,使用CD-HIT程序排除相似度大于20 %的序列。**这种方法最大限度地减少了序列之间的相似性,从而能够更准确地识别和预测不同亚细胞位置的序列。因此,获得了四个部分的数据集:细胞核,细胞质,核糖体和细胞质。如图1所示,这些数据集显示了每个亚细胞定位的序列数量和序列长度的分布。
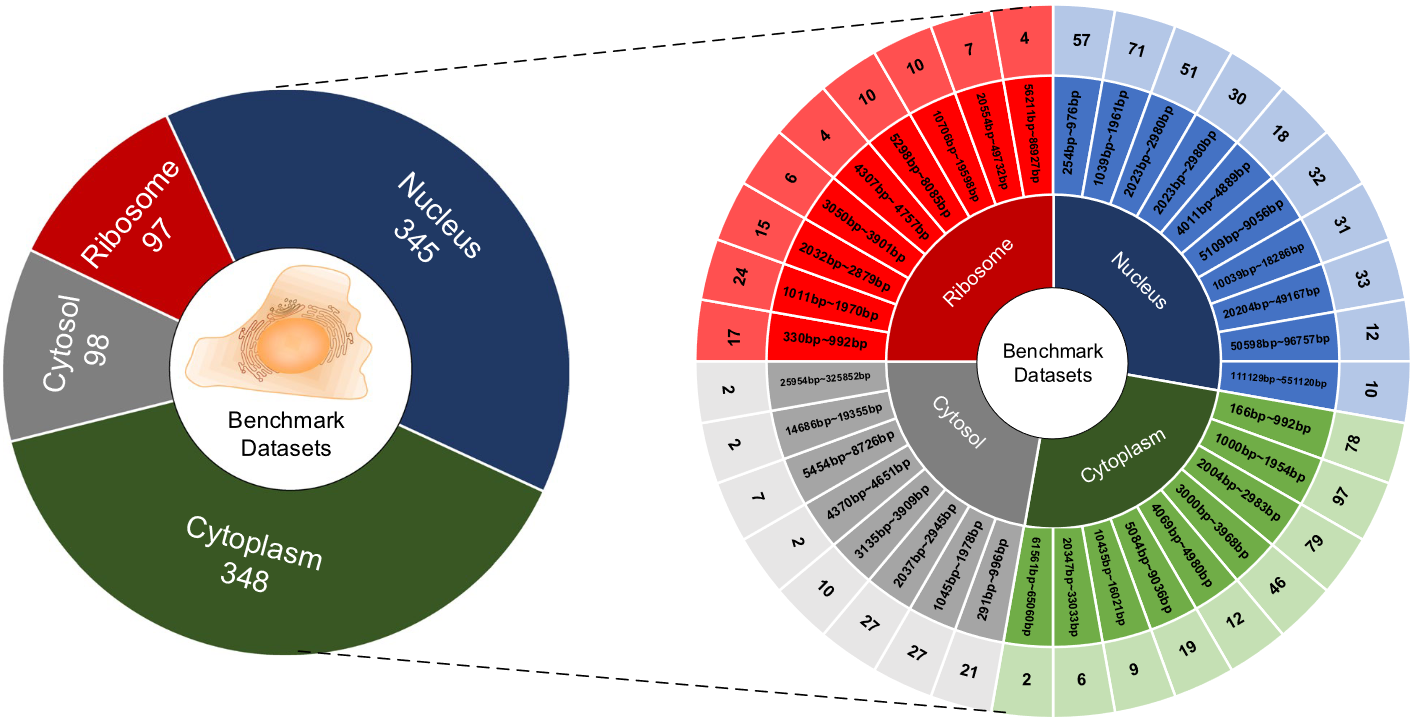 图1数据集中定位于每个子细胞的lncRNA数量分布
图1数据集中定位于每个子细胞的lncRNA数量分布
为了实验和评估的目的,每个类别从数据集中随机划分,其中20 %作为独立的测试集,其余部分作为训练集。在整个训练过程中,从训练集中提取一个验证集来微调模型的超参数并检测潜在的过拟合。在模型训练完成后,使用测试集对模型的性能进行综合评估。基准数据集的设置可以概括如下:
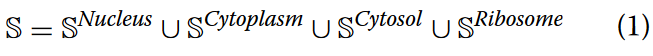
多类修改的核苷酸位置感知编码
深度学习模型由于其固有的对数值输入的依赖,通常不能直接处理原始基因序列。为了将序列中的四种基本核苷酸组成转换为适合模型学习的数值格式,大量的序列编码方法被提出。其中比较流行的编码有One-hot编码、编码的核酸化学性质(Nucleic acid Chemical Properties,NCP)和编码的二核苷酸物理化学性质(Dinucleotide Physical and Chemical Properties,DPCP) 。这些方法基于人工定义的规则将基因序列编码为数值矩阵,显著提升了模型学习的效率和性能。然而,它们仍然不能全面地捕获核苷酸在序列中的位置特征 。为了生成更全面的序列数值表示,提取lncRNA序列内部不同位置核苷酸的分布模式进行亚细胞定位,更好地解决亚细胞定位的多类问题,提出了基于多类修正核苷酸密度的归一化差异位置感知K-mer编码方法(MCD-ND)。
为了将DNA序列转换为数值表示,首先需要使用长度为k的固定大小窗口和指定的步长将其分割成k-mers 。每个片段,即k-mers,代表一组不同的核苷酸。这些k-mers的大小取决于用于分割的窗口大小。收集所有唯一的k-mers组成一个词汇表,词汇表的大小由k的值决定,假设有足够数量的序列样本。例如,当k=1时,词汇由4个可能的k-mers组成,分别对应于核苷酸A、C、G和T;当k=2时,词汇扩展到16个不同的k-mers,包括AA,AC,AG,AT等。生成的k-mers在每个序列中的位置可以表示为Pi = P1,P2 · · ·。随着k-mers的大小增加,词汇表的大小也随之增加,并且随着序列的变长,位置的数量也相应增加。
将序列切分为k-mers后,分别统计各类别序列中不同位置词汇的出现频率。该过程产生多个矩阵,标记为Aclassc,每个矩阵具有z位置的维度和n个词汇项,如公式2所示。
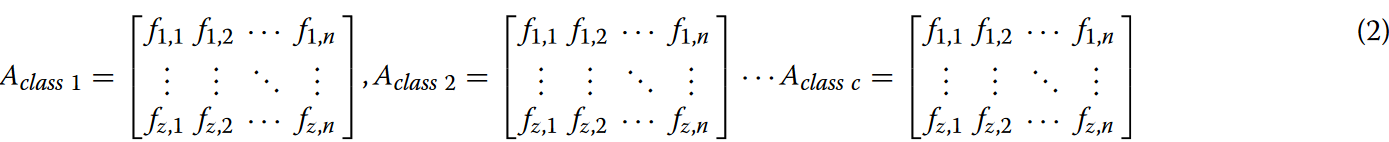 矩阵中的每个元素fi,j表示序列中第j个词在第i个位置的出现频率,
矩阵中的每个元素fi,j表示序列中第j个词在第i个位置的出现频率,
其中c表示类别数,表示不同类型的基因序列。
在获得每个序列类别的频率分布矩阵后,进一步统计序列中k-mers的总数 ,记为NSc。对频率分布矩阵进行归一化处理,得到其密度分布矩阵Aden classc,如公式3所示。
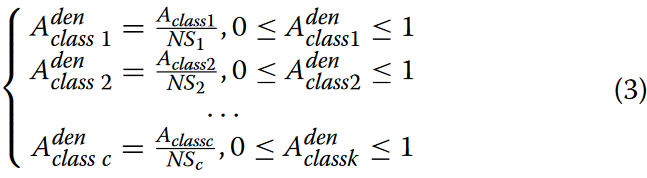
为了实现对DNA序列更全面的统计表示,我们利用每个类别的密度分布矩阵计算基因序列第i个位置的第j个k-mer的位移传感器Specific三核苷酸Propensity(PSTNPss)得分。式(4)给出了计算PSTNPss得分的一般数学表达式。
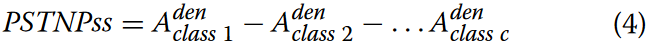
不同类别的k-mers在相同位置的密度分布往往表现出差异,在某些情况下,这种差异是显著的。为了增强PSTNPss得分的判别能力,引入类别平衡因子,得到MCD-ND得分矩阵,如公式5所示。
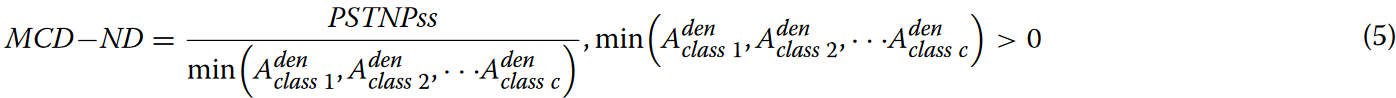
长度为L的基因序列被分割成L-k+1个k-mers。然后根据这些k-mers在序列中的顺序位置,基于MCD-ND得分矩阵,将这些k-mers编码成1×(L-k+1)矩阵。随后,将此矩阵输入到模型中进行学习。当class=2,k=2时,基因序列的编码过程如图2所示。
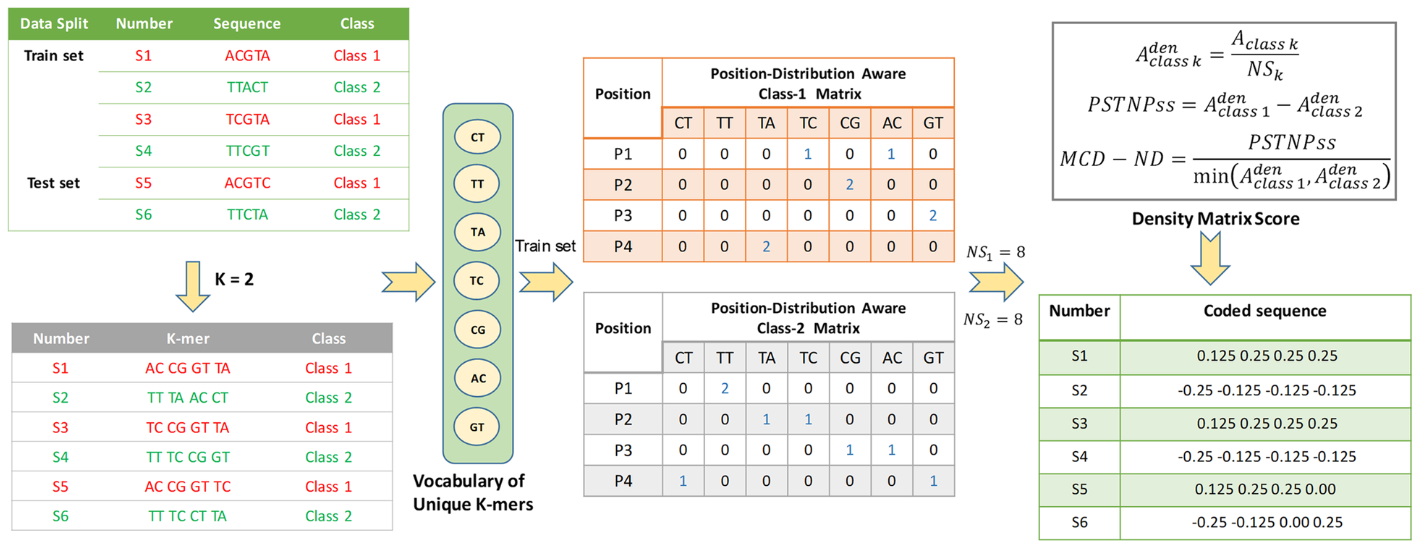 图2 MCD-ND编码流程示例
图2 MCD-ND编码流程示例
构建模型
在将亚细胞定位的lncRNA序列编码为机器学习模型所需的数值矩阵后,我们利用各种深度学习神经网络( deep learning neural network,DNN )算法构建多类预测因子,旨在揭示包含核苷酸特定位置分布的特征矩阵中的隐藏信息。图3说明了用于亚细胞定位的多类预测器的构造。该模型主要包括四个部分,统称为MGBLncLoc。第一部分构成特征增强模块,该模块结合了Multi Dconv Head Transposition Attention (MDTA)模块和门控Dconv前馈网络(GDFN)模块 。第二部分包括多尺度卷积神经网络(CNN)模块 。第三部分涉及双向门控循环单元(Bidirectional Gated Recurrent Unit,BiGRU)模块,负责处理上下文相关序列 。最后,由全连接层组成的分类模块用于对前面网络层提取的信息进行非线性处理。
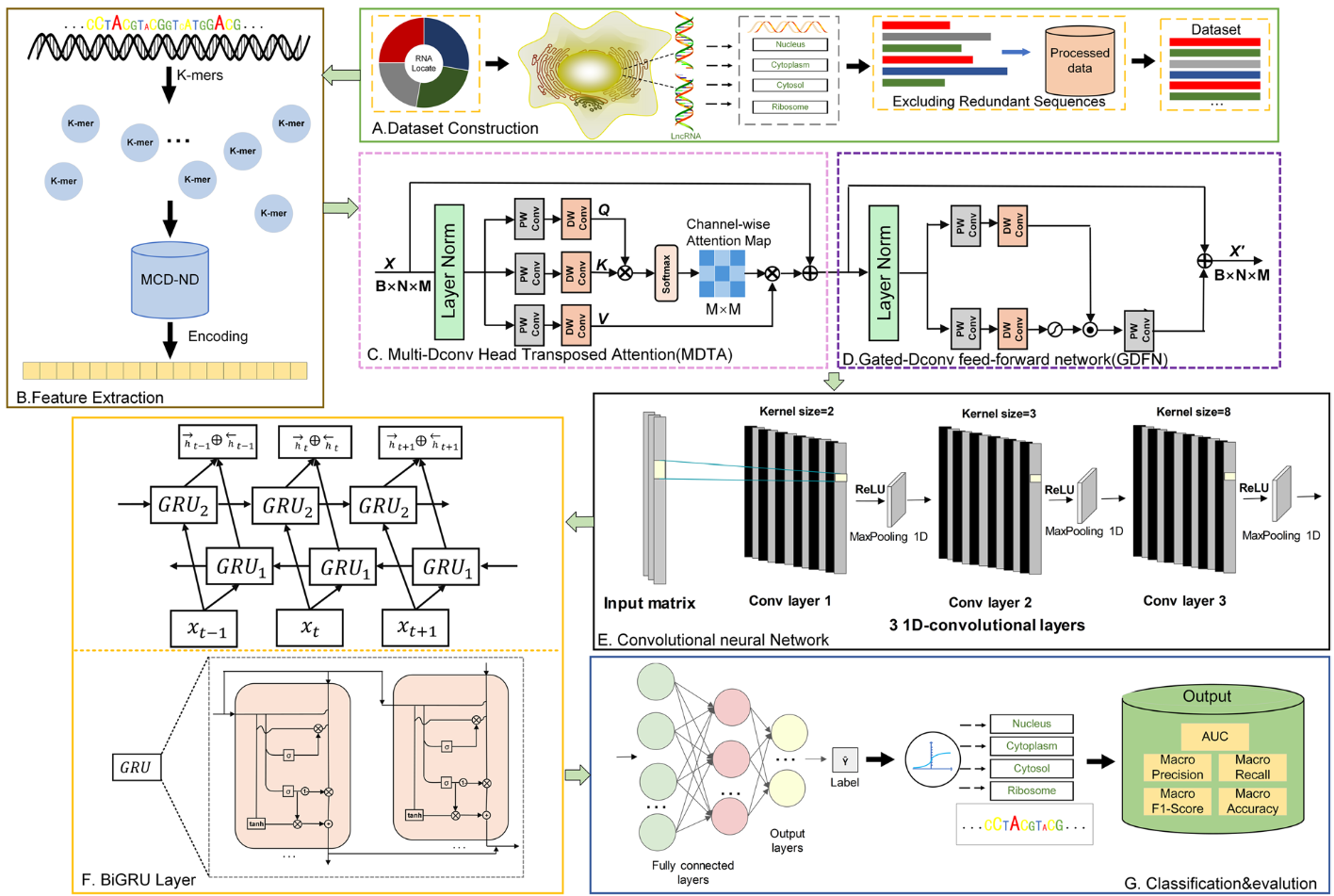 图3 模型MGBLncLoc的工作框架
图3 模型MGBLncLoc的工作框架
为了有效融合从编码序列中提取的特征信息,我们设计了基于通道注意力的特征增强模块 。该模块首先计算通道注意力 ,进一步巩固特征信息。随后,融合后的特征通道通过重建层降为奇异通道 ,得到最终的增强特征。这种设计有利于模型全面地捕获序列数据内部的特征关系,从而生成更具表达能力的特征表示,增强模型的性能。该模块包括两个部分:MDTA模块和GDFN模块 。为了减轻网络的计算负担,MDTA计算跨信道的互协方差 。首先,使用逐点卷积(PW Conv)和深度卷积(DW Conv)对输入特征Mi进行处理 ,得到Q∈B×N×M,K∈B×N×M,V∈B×N×M。PW Conv对信道进行内容编码并整合信道间的上下文信息 ,而DW Conv则进一步对空间上下文进行编码 。随后,进行重塑操作,得到Q∈B×N×M,K∈B×N×M,V∈B×N×M。计算ΔQ与ΔK的点积,生成大小为M×M的通道注意力图A,如式(6)所示。

在得到注意力图A后,我们对V进行加权操作,将其与A相乘得到增强后的特征表示。具体操作如下:

这里,V′表示加权特征图,增强通道间关系。随后,通过重构和加性残差连接,将这些特征图无缝集成到初始特征中 。为了得到更精确的残差信息,我们使用GDFN进行复杂的操作。最初,输入特征经过深度卷积编码空间上下文信息。这里,V′表示加权特征图,增强通道间关系。随后,通过重构和加性残差连接,将这些特征图无缝集成到初始特征中。为了得到更精确的残差信息,我们使用GDFN进行复杂的操作。最初,输入特征经过深度卷积编码空间上下文信息。

其中,φ(x)为标准正态分布的累积分布函数(CDF),通常计算为:
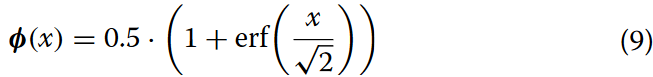
在这一部分中,MDTA模块主要用于捕获序列数据中的局部特征,并通过注意力机制强调不同位置之间的关系 。结合多传感头可分解卷积和转置注意力机制,帮助模型更好地理解输入序列中的局部结构。同时,GDFN模块集成了门控卷积和前馈神经网络的结构 。门控卷积通常用于控制信息的流动,而前馈神经网络则处理特征表示。GDFN的主要作用是更有效地处理序列数据。通过门机制,它调节信息流,帮助模型更有效地捕获序列数据中的全局特征。
在CNN模块中,卷积层执行类似于使用滑动窗口的操作,从具有高激活特征信息的序列中提取模体 。因此,MGBLncLoc利用三个1D卷积层,在特征矩阵上用多个卷积块并行地进行卷积。特征向量通过多尺度卷积层导出 ,以修正线性单元( ReLU )作为激活函数。在获得卷积特征矩阵后,应用最大池化通过减少特征数量来减轻过拟合。卷积层的数学表示如式(10)所示。
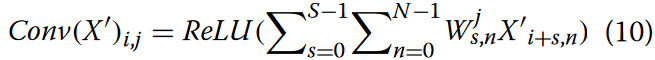
在该方程中,A表示对基因序列编码后得到的增强特征矩阵 ,其中i为输出位置的索引,j为滤波器的索引。每个卷积滤波器Wj是一个S×N的矩阵,其中S为超参数优化确定的滤波器尺寸,N为输入通道数。对于第一个卷积层,N是序列编码后特征矩阵的输入维度。ReLU表示为:

目前,大多数基因序列数据的处理依赖于循环神经网络架构,如LSTM、GRU等。其中BiGRU在分类任务中取得了显著的成功。BiGRU在序列的前向和后向两个方向上都能捕获时间序列特征,相比于单独的GRU,产生了更加鲁棒和信息丰富的特征表示。在MGBLncLoc模型中,BiGRU利用其内部状态来处理序列向量,充分利用了两个方向的序列上下文信息。门控循环单元( Gated Recurrent Units,GRUs )构成了BiGRUs的主要组成部分,用于动态记忆或遗忘序列信息。GRU层的主要操作包括更新门和复位门。更新门控制新的输入信息与前一个隐藏状态的融合程度,而重置门则调节前一个隐藏状态如何用于计算候选激活值。这些门的计算涉及到输入和前一个隐藏状态的权重矩阵的线性组合,由Sigmoid函数激活。通过这些门的集成,BiGRU有效地控制了信息的流动和保留。具体来说,第j个隐藏单元在时间步t的更新门zt j和重置门rt j计算如方程12所示。
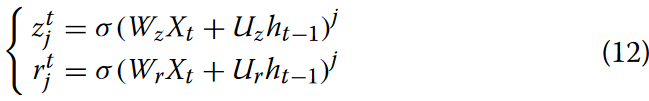
式中,σ表示逻辑sigmoid函数,Wz和Uz表示学习到的不同权重矩阵,ht-1表示之前的隐藏状态。Xt表示输入,其中在第一个单元中,多尺度卷积网络层的输出作为输入。
在更新和重置门之后,根据公式11计算第j个隐藏单元在时间步长t的激活值ht j。其中候选激活值'~ht j是通过将双曲正切函数tanh应用于输入数据和先前隐藏状态的组合来确定的。
BiGRU以这种方式在序列数据上动态更新隐藏状态,以更好地捕获序列内部的重要特征和关系。这种双向处理使模型能够更全面地理解序列数据,从而提高模型在各种序列任务上的性能。
在分类模块中,前一层网络层提取的信息通过全连接层进行非线性处理。这些全连接层的输出向量作为整流线性单元( ReLU )激活函数的输入。在多类亚细胞定位问题的背景下,每个节点的概率是独立的,导致每个节点的得分从0到1不等。最终,MGBLncLoc试图建立以下映射关系:
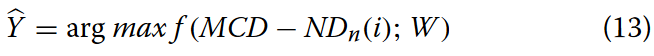
式中:^Y为lncRNA序列亚细胞定位的神经网络预测得分;MCD-NDn表示经过MCD-ND编码的序列的特征矩阵;W表示神经网络的参数;而f是神经网络所寻求的映射函数。
为了建立这种映射关系,需要定义一个损失函数来量化预测标签和真实标签之间的差异。我们采用了常用的交叉熵损失函数,如公式14所示。
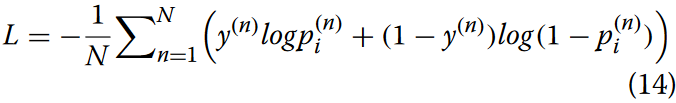
其中N表示样本容量,y(n)为二分类变量,p(n)i为神经网络对第i个亚细胞定位的第n个样本的预测概率。
模型性能评估
为了评估机器学习模型的分类性能,采用了几种广泛用于多类分类问题的评估指标,遵循了先前研究中使用的方法。这些评价指标包括宏观精度( Macro- Precision )、宏观召回率( Macro- Recall )、宏观F1分数( Macro- F1- Score )和宏观准确度( Macro- Precision ),并结合ROC曲线下面积( Area Under the ROC Curve,AUC )进行综合评价。首先,计算每个亚细胞定位类别的精确率、召回率和准确率,然后进行宏平均计算。下面概述每个评价指标的计算过程。
 其中,N表示类别总数,TPi表示类别i的真阳性个数,TNi表示类别i的真阴性个数,
其中,N表示类别总数,TPi表示类别i的真阳性个数,TNi表示类别i的真阴性个数,
FPi表示类别i的假阳性个数,FNi表示类别i的假阴性个数。
AUC定义为ROC曲线与坐标轴围成的面积。AUC值越高,越接近1.0,表明模型性能越好。
结果
不同特征编码方式对模型性能的影响
用于编码序列特征的方法也直接影响模型的性能。不同的特征编码方式将序列转化为不同的特征表示,辅助机器学习模型处理基因序列。在生物信息学研究中,通常采用不同的编码方式。其中包括One-hot编码、基于核苷酸化学性质的NCP编码、基于二核苷酸化学性质的DPCP编码、基于K-mer频率的编码方法以及Word2Vec等自然语言处理编码技术。由于这些方法在表示生物序列方面的有效性,近年来得到了广泛的应用。与MCD-ND编码类似,这些方法涉及手工定义的规则,将序列表示为不同的特征。因此,我们重新生成这些特征作为模型的输入,并与3-mer MCD-ND编码进行比较,以证明MCD-ND编码的有效性。为了获得更可靠的实验结果,每组模型训练了十次,并计算了每个指标的平均值,并列在表2中。
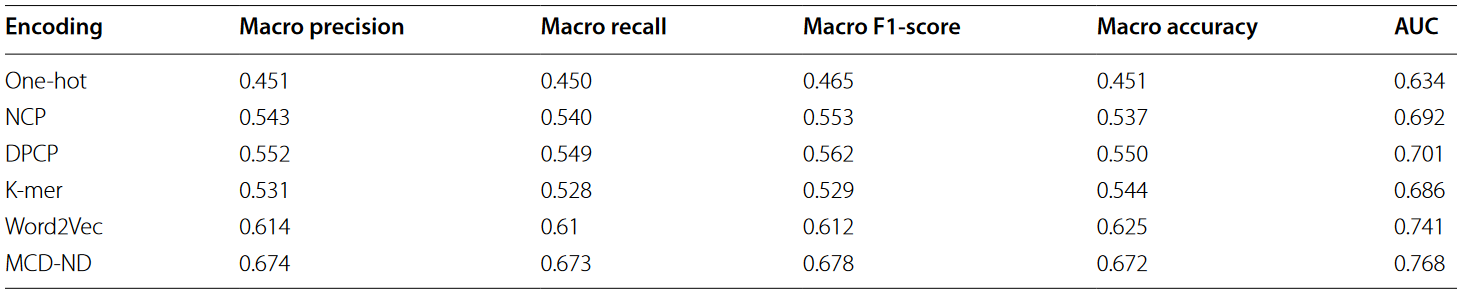 表2 亚细胞定位模型识别的平均值
表2 亚细胞定位模型识别的平均值
与现有亚细胞定位预测模型的比较
不同的数据集会导致模型识别结果的差异。lncLocator、iLocLncRNA、Locate-R、DeepLncLoc、GraphLncLoc等模型均使用RNALocate v1.0数据库进行训练和测试。为了确保公平的比较并证明MGBLncLoc模型的优越性,使用RNALocate v1.0对其进行了重新训练,以便进行比较。与RNALocate v2.0不同的是,v1.0数据集中位于细胞质中的lncRNA序列数量略多于位于细胞核中的lncRNA序列数量,这表明两个数据集之间存在明显的分布模式。这种比较得到的评价结果如图7所示。
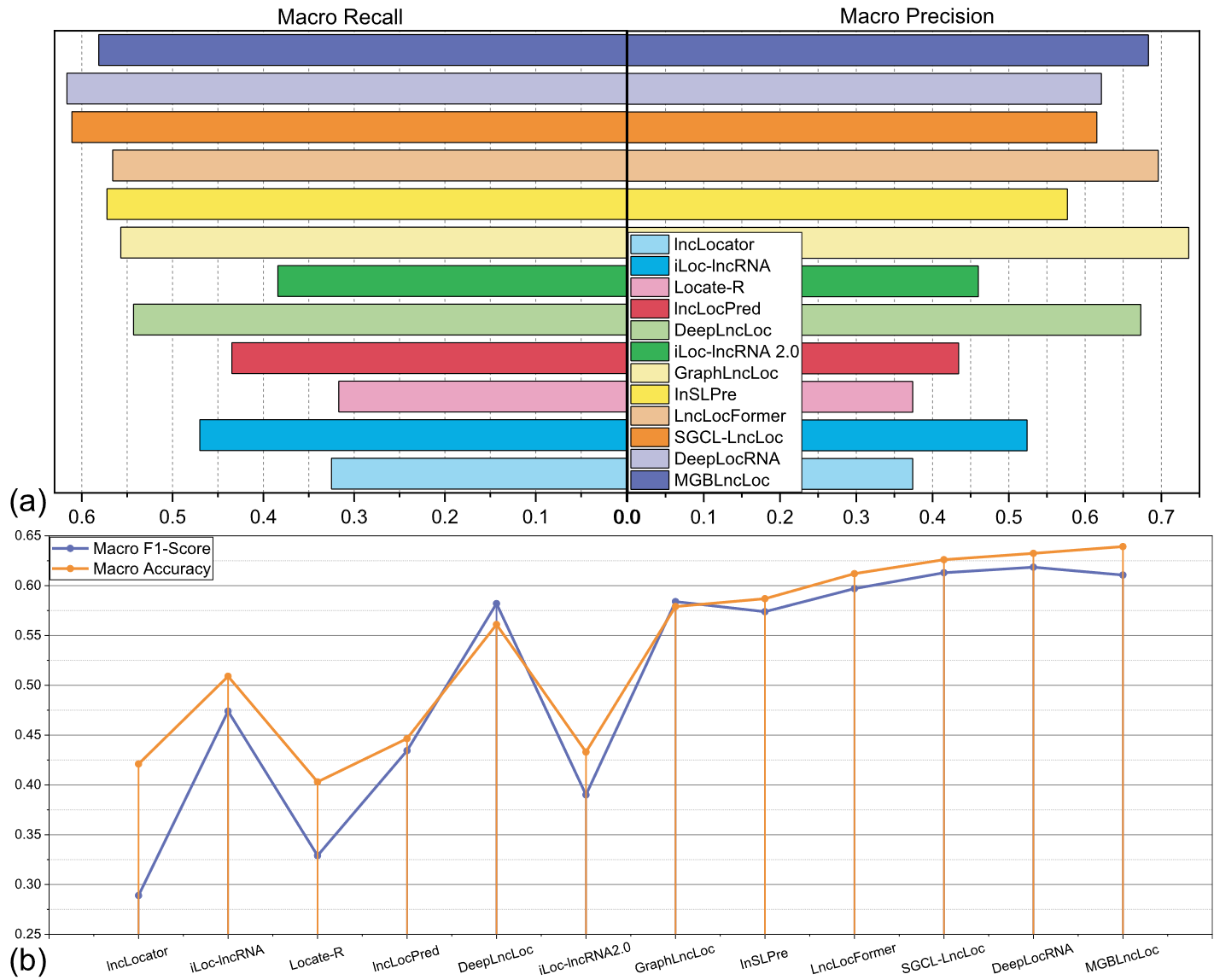 图7各对比模型实验的评价结果。
图7各对比模型实验的评价结果。
a各对比模型的宏查准率和宏查全率的评价结果。b各对比模型的宏F1分数和宏查准率的评价结果
在图7a中,MGBLncLoc模型在宏平均召回率方面优于相对较高的模型LncLocFormer,lncSLPre,SGCLLncLoc和DeepLocRNA。图7b表明,MGBLncLoc取得了宏平均F1分数的最高评价,达到了61.1 %。F1分数代表精确率和召回率的调和平均值,表示模型对每个亚细胞定位类别的分类精度和识别能力。此外,MGBLncLoc的宏平均准确率最高,达到63.5 %,超过其他亚细胞预测模型,取得了较好的效果。