做了微生物检测,得到一堆数据,今天开始挑着讲一下其中的LefSE分析和LDA分析。在宏基因组分析中,LEfSe分析 和LDA分析 并不是两个完全独立的"东西",而是密切相关、属于同一分析流程中的不同环节。你可能会在结果中看到类似下面类似的两张图:
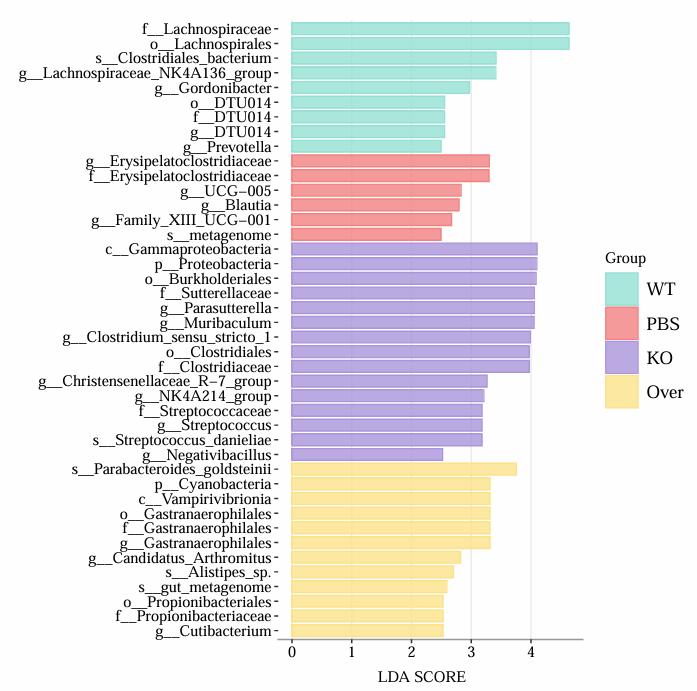
图1 LDA score 图
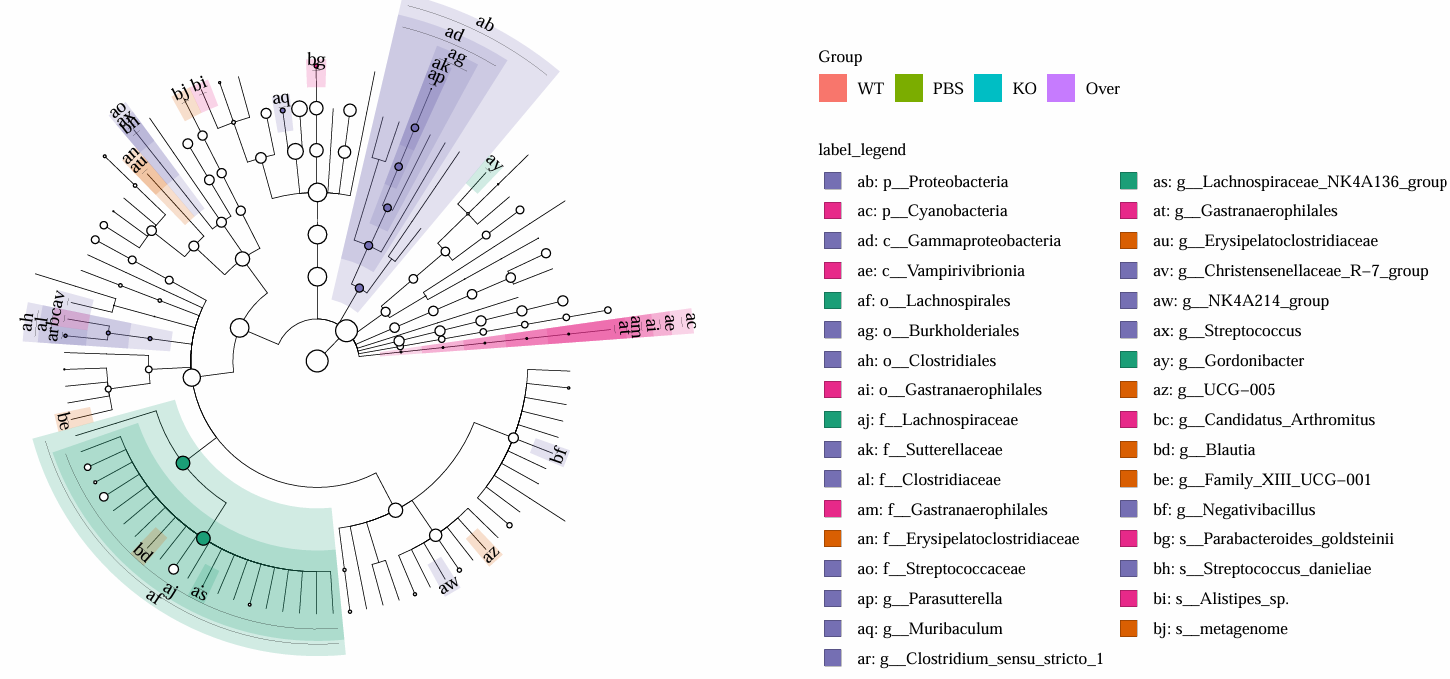
图2 Cladogram图
✅ 图1:分类单元柱状图(LDA score 图)
-
横轴:LDA score(对数尺度,通常 >2 或 >3 才显著)
-
纵轴:差异显著的分类单元(从门到种)
-
颜色:不同颜色代表不同分组(如 WT、PBS、KO、Over)
-
怎么看?
-
LDA值越大 → 该菌在该组中越特异 、贡献越大
-
只在某一侧出现 → 是该组的biomarker
-
左右都有 → 可能在多组中都有差异,需结合图2看
-
✅ 图2:进化分支图(Cladogram)
-
圆圈 :从外到内依次是 种 → 属 → 科 → 目 → 纲 → 门
-
颜色:不同颜色代表不同分组(如红色是KO,绿色是WT)
-
怎么看?
-
黄色节点:差异不显著
-
彩色节点:该分类单元在该组中显著富集
-
节点大小:LDA值越大,节点越大
-
🔍 举个例子(从图中可以看到):
✅ 图1中看到的:
-
g_Lachnospiraceae_NK4A136_group在 WT组 中显著富集(LDA ≈ 4.5) -
g_Streptococcus在 Over组 中显著富集(LDA ≈ 4.0) -
s_Parabacteroides_goldsteinii在 KO组 中显著富集(LDA ≈ 3.8)
✅ 图2中看到的:
-
外圈(种水平):
s_Parabacteroides_goldsteinii节点为 红色 → 在 KO组 显著富集
-
中圈(属水平):
g_Streptococcus节点为 蓝色 → 在 Over组 显著富集
✅ 总结一句话:
看谁在哪个组"特别多",LDA值越大、颜色越纯,越可能是该组的标志菌。
✅ 简明结论:
LEfSe 是一种完整的差异分析流程,LDA 是这个流程中的最后一步。
🔍 分析详细解释:
- LEfSe(Linear Discriminant Analysis Effect Size)
LEfSe 是一种用于发现组间显著差异特征(如微生物物种、功能基因等)的分析方法,常用于宏基因组、16S rRNA 等微生物组数据。它整合了统计检验 和效应量评估 ,强调统计显著性 与生物学一致性。
LEfSe 分析流程包括三个步骤:
-
Kruskal-Wallis 秩和检验:筛选在不同组间丰度差异显著的物种;
-
Wilcoxon 秩和检验(如有亚组):检验这些差异是否在各亚组间一致;
-
线性判别分析(LDA):评估每个差异物种的效应大小(即 LDA score),用于排序和可视化。
-
LDA(Linear Discriminant Analysis)
LDA 是一种降维和分类方法,在 LEfSe 中用于:
-
计算每个差异特征(如物种)的效应值(LDA score);
-
判断其对组间差异的贡献度;
-
提供可视化依据(如 LDA 值柱状图、系统进化树)
分析
数据
见附件资源,也可以callme发小伙伴,反正是随机弄个出来做测试的。
R脚本
R
getwd()
rm(list=ls()) #清空工作环境
# 安装必要的包(如果尚未安装)
# install.packages(c("tidyverse", "microeco", "magrittr", "openxlsx"))
library(tidyverse)
library(microeco)
library(magrittr)
library(openxlsx)
# 加载系统发育树相关包
library(BiocManager)
# BiocManager::install("ggtree") # 如未安装请取消注释
library(ggtree)
list.files()
# 读取数据
feature_table <- read.table('feature_table.txt', header = TRUE, row.names = 1)
sample_table <- read.table('sample_table.txt', header = TRUE, row.names = 1)
tax_table <- read.table('tax_table.txt',
header = TRUE,
row.names = 1,
fill = TRUE) # 短行补 NA
colnames(tax_table) <- c("Kingdom", "Phylum", "Class", "Order",
"Family", "Genus", "Species")
# 手动计算相对丰度并转换为data.frame(关键修改)
feature_table_relative <- as.data.frame(t(t(feature_table) / colSums(feature_table, na.rm = TRUE)) * 100)
# 确保行名正确保留
rownames(feature_table_relative) <- rownames(feature_table)
# 创建microtable对象,使用转换后的data.frame格式
dataset <- microtable$new(sample_table = sample_table,
otu_table = feature_table_relative,
tax_table = tax_table)
# 整理分类学信息,添加标准前缀(k__、p__等)
dataset <- tidy_taxonomy(dataset)
# 执行LEfSe分析
lefse <- trans_diff$new(dataset = dataset,
method = "lefse",
group = "Group",
alpha = 0.05,
taxa_level = "all",
lefse_subgroup = NULL)
# 查看和保存结果
View(lefse$res_diff[lefse$res_diff$LDA > 2, ])
# dir.create("WT-PBS-KO-Over") # 如未创建目录请取消注释
write.xlsx(lefse$res_diff[lefse$res_diff$LDA > 2, ], file = "WT-PBS-KO-Over/lefse_result.xlsx")
# 绘制柱状图
index <- which(lefse$res_diff$LDA > 2)
color_map <- c("WT" = "#95e1d3", "PBS" = "#f38181", "KO" = "#aa96da", "Over" = "#fce38a")
plot <- lefse$plot_diff_bar(use_number = index,
width = 0.8,
alpha = 0.8,
group_order = c("WT", "PBS", "KO", "Over"))
plot <- plot + scale_color_manual(values = color_map) +
scale_fill_manual(values = color_map) +
# ggtitle("Biomarker (Genus)") +
# theme(plot.title = element_text(hjust = 0.46, vjust = 0, size = 23)) +
ylab("LDA SCORE") +
theme(axis.title.x = element_text(hjust = 0.46, vjust = -1, size = 12, color = "black"),
axis.text.x = element_text(size = 12, colour = "black")) +
theme(axis.text.y = element_text(size = 12, color = "black")) +
theme(text = element_text(size = 12, color = "black")) +
theme(legend.position = "right",
legend.justification = c(0.7, 0.5),
legend.direction = "vertical",
legend.text = element_text(size = 14),
legend.key.size = unit(0.9, "cm")) +
theme(plot.margin = margin(2, 1, 1, 1, "cm")) +
theme(text = element_text(family = "serif"))
print(plot)
ggsave(filename = "WT-PBS-KO-Over/WT-PBS-KO-Over.barplot.png", plot = plot, width = 8, height = 8, dpi = 300)
ggsave(filename = "WT-PBS-KO-Over/WT-PBS-KO-Over.barplot.pdf", plot = plot, width = 8, height = 8)
dev.off()
# 绘制系统进化树图
treeplot <- lefse$plot_diff_cladogram(
use_taxa_num = 200,
use_feature_num = 50,
clade_label_level = 7,
group_order = c("WT","PBS","KO","Over")
)
# 1. 修改进化树标签字体和大小,避免重叠
for (i in seq_along(treeplot$layers)) {
if (any(c("GeomText", "GeomLabel") %in% class(treeplot$layers[[i]]$geom))) {
# 适当调整字号,添加文字间距参数
treeplot$layers[[i]]$aes_params <- modifyList(
treeplot$layers[[i]]$aes_params,
list(
family = "serif",
size = 4, # 适度减小字号避免重叠
hjust = 0.5, # 水平居中
vjust = 0.5, # 垂直居中
lineheight = 0.9 # 调整行高
)
)
}
}
# 2. 添加其他设置,增大进化树显示空间
treeplot <- treeplot +
# 拆分图例
guides(
color = guide_legend(
title = "Biomarker",
order = 1,
position = "right",
keywidth = 0.8, # 减小图例宽度
keyheight = 0.8
),
fill = guide_legend(
title = "Group",
order = 2,
position = "right",
nrow = 1 # 水平排列底部图例
)
) +
# 主题字体设置
theme(
text = element_text(family = "serif"),
axis.text = element_text(family = "serif"),
legend.text = element_text(family = "serif", size = 10), # 减小图例文字
legend.title = element_text(family = "serif", size = 10),
# 调整边距,为进化树腾出更多空间
plot.margin = margin(0.5, 0.5, 0.5, 0.5, "cm"),
# 移除不必要的元素(如果存在)
axis.title = element_blank(),
axis.ticks = element_blank()
)
# 3. 显示时使用更大的绘图设备
# 新建一个更大的绘图窗口
dev.new(width = 18, height = 6) # 增大宽高比例,适合树状图
print(treeplot)
# 保存进化树图
ggsave(filename = "WT-PBS-KO-Over/WT-PBS-KO-Over.cladogram.png", plot = treeplot, width = 18, height = 8, dpi = 300)
ggsave(filename = "WT-PBS-KO-Over/WT-PBS-KO-Over.cladogram.pdf", plot = treeplot, width = 14, height = 8)