文章目录
- 二,结构域数据库的API接口:InterPro为例
-
- 1,CATH-Gene3D(https://www.cathdb.info/(https://www.cathdb.info/))
- 2,CDD(https://www.ncbi.nlm.nih.gov/cdd/(https://www.ncbi.nlm.nih.gov/cdd/))
- 3,HAMAP(https://hamap.expasy.org/(https://hamap.expasy.org/))
- 4,NCBIFAM(https://www.ncbi.nlm.nih.gov/genome/annotation_prok/evidence/(https://www.ncbi.nlm.nih.gov/genome/annotation_prok/evidence/))
-
- [1. 隐马尔可夫模型(HMMs)](#1. 隐马尔可夫模型(HMMs))
- [2. BlastRules](#2. BlastRules)
- [3. 结构域架构](#3. 结构域架构)
- 5,PANTHER(https://pantherdb.org/(https://pantherdb.org/))
- 6,PIRSF(https://proteininformationresource.org/pirsf/(https://proteininformationresource.org/pirsf/))
- 7,PRINTS(https://www.ebi.ac.uk/interpro/entry/prints/)(https://interpro-documentation.readthedocs.io/en/latest/prints.html(https://interpro-documentation.readthedocs.io/en/latest/prints.html))
- 8,Pfam(https://pfam.xfam.org/(https://pfam.xfam.org/))
- [9,PROSITE profiles/PROSITE patterns(https://www.ebi.ac.uk/interpro/entry/prosite/)(https://prosite.expasy.org/(https://prosite.expasy.org/))](#9,PROSITE profiles/PROSITE patterns(https://prosite.expasy.org/))
- 10,SFLD(http://sfld.rbvi.ucsf.edu/archive/django/index.html(http://sfld.rbvi.ucsf.edu/archive/django/index.html))
- 11,SMART(https://smart.embl.de/(https://smart.embl.de/))
- 12,SUPERFAMILY(https://www.ebi.ac.uk/interpro/entry/ssf/)(https://supfam.org/SUPERFAMILY/(https://supfam.org/SUPERFAMILY/))
- 13,总结:从InterPro出发
-
- 重点:如何使用&接口访问
-
- (1)InterProScan软件:
- [(2)InterPro API(应用程序编程接口)](#(2)InterPro API(应用程序编程接口))
- (3)官方调用API的脚本生成器
- 三,to-do
本篇是系列2,系列1也就是上一篇文章,参考链接:https://blog.csdn.net/weixin_62528784/article/details/153889499?sharetype=blogdetail&sharerId=153889499&sharerefer=PC&sharesource=weixin_62528784&spm=1011.2480.3001.8118
二,结构域数据库的API接口:InterPro为例
正如上一个section所述,我们不好从unirprot这种综合性数据库去获取注释(当然,你当然可以只用uniprot数据库,实际使用或者写文献都可以,只是我个人认为不严谨),究其原因是因为综合性蛋白质数据库是一个大杂烩,一般我们第一次接触的就是这种数据库,但是信息太杂,其实不好统一,
我宁愿要一个统一地rule、结构域识别规则,
而不是在文献中说"unirprot数据库获取结构域数据",毕竟uniprot只是收集数据,
结构域的注释有其rule规则,但是这个规则本身又不是uniprot数据库的。
我个人是倾向于用统一的数据注释rule,来规范我的数据来源的。
如section1中所述,其实我们一般所用的结构域注释数据库,也就那么几个:

有些并不单纯是结构域的,但是我还是统一归纳进来,最后一个MobiDB是无序区域数据库,因为此处我们是要找domain,不是IDR,所以不将其列入在内。
另外再补充一点Interpro数据库的成员子数据库,都是一些和蛋白质相关的:
参考https://www.ebi.ac.uk/interpro/,


其实一般用的多的,就是Interpro数据库和Pfam数据库,因为Pfam数据库已经归档到Interpro数据库里了,
所以我们按照Interpro的组成成员数据库(13个成员数据库,但是我们只对其中涉及到结构域的数据库进行分析)的顺序来展开介绍。
粗略归纳:
plain
CATH-Gene3D
CDD
HAMAP
NCBIFAM
PANTHER
Pfam
PIRSF
PRINTS
PROSITE profiles
PROSITE patterns
SFLD
SMART
SUPERFAMILY其实大部分蛋白质数据库,我之前的博客都涉及到。
具体查询的时候,可以看一下数据库所被cite的文献,可以了解一下数据库具体是做什么的,具体更新了什么数据(某种程度上,看数据库发表的文献,和看数据库官方文档,其实从高屋建瓴概览的角度效果是一致的)
具体参考:
https://interpro-documentation.readthedocs.io/en/latest/databases.html

1,CATH-Gene3D(https://www.cathdb.info/)


本质是结构域分类的数据库:
Gene3D是基于蛋白质序列的结构域预测数据库,通过利用CATH分类的信息,使用序列比对和HMMs来预测蛋白质序列中的结构域。
2,CDD(https://www.ncbi.nlm.nih.gov/cdd/)


结构域注释以搜索的数据库:具体的一些条目细节
参考:https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml



快速入门指南参考:https://www.ncbi.nlm.nih.gov/Structure/cdd/docs/cdd_how_to.html
常见类型的搜索、常见问题:


重点:如何使用&接口访问

(1)单个序列:
使用CD-Search:

https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
此处我还是以CTCF为例,我输入的是序列,并非任何ID,结构域注释结果界面主要内容如下:
可以发现,注释还是从推断序列的相似物身份开始的,
所以基本上所谓的注释,还是先从序列数据库搜索开始(类似Blast那一套),再给出高相似性序列hit的注释数据,
这一点对于有名有姓的蛋白质序列数据比较友好

这个NP_006556其实就是CTCF_human的"有名有姓"的注释
https://www.ncbi.nlm.nih.gov/protein/NP_006556
所以这个页面中接下来的结构域注释,其实就是直接调用CTCF这个"有名有姓"的蛋白质的数据了(再次强调,我是仅输入CTCF蛋白质序列,并没有给出ID提示,这一点在写脚本时比较重要-直接基于序列数据)
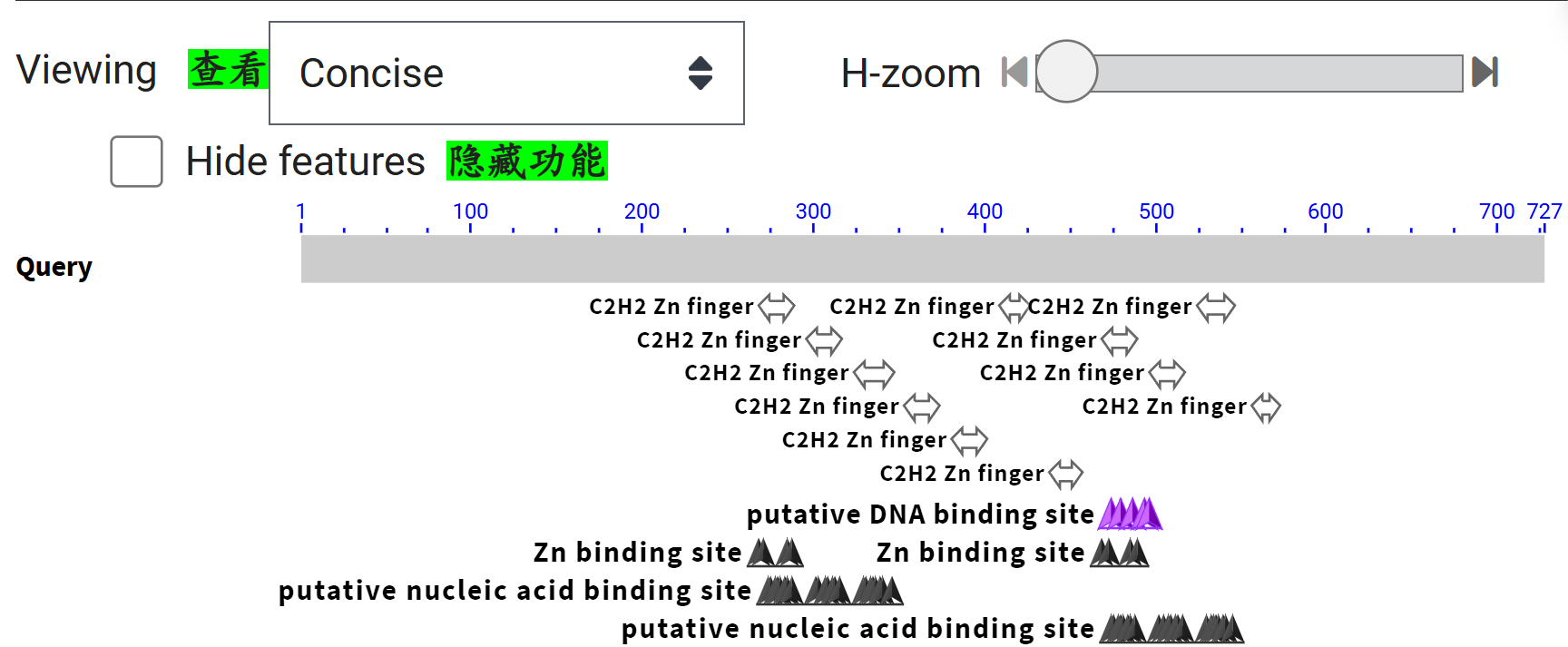
可以看到,有11个ZF结构域,和前面Uniprot中
(2)多个序列:
使用Batch CD-Search,

web server界面参考:https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi
自动化脚本下载:
如果要整合到数据分析流程中,也就是python pipeline中,也就是脚本化下载,本博客的重点,
参考:https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml#BatchRPSBWebAPI

按照官方提示,我们主要使用HTTP POST request
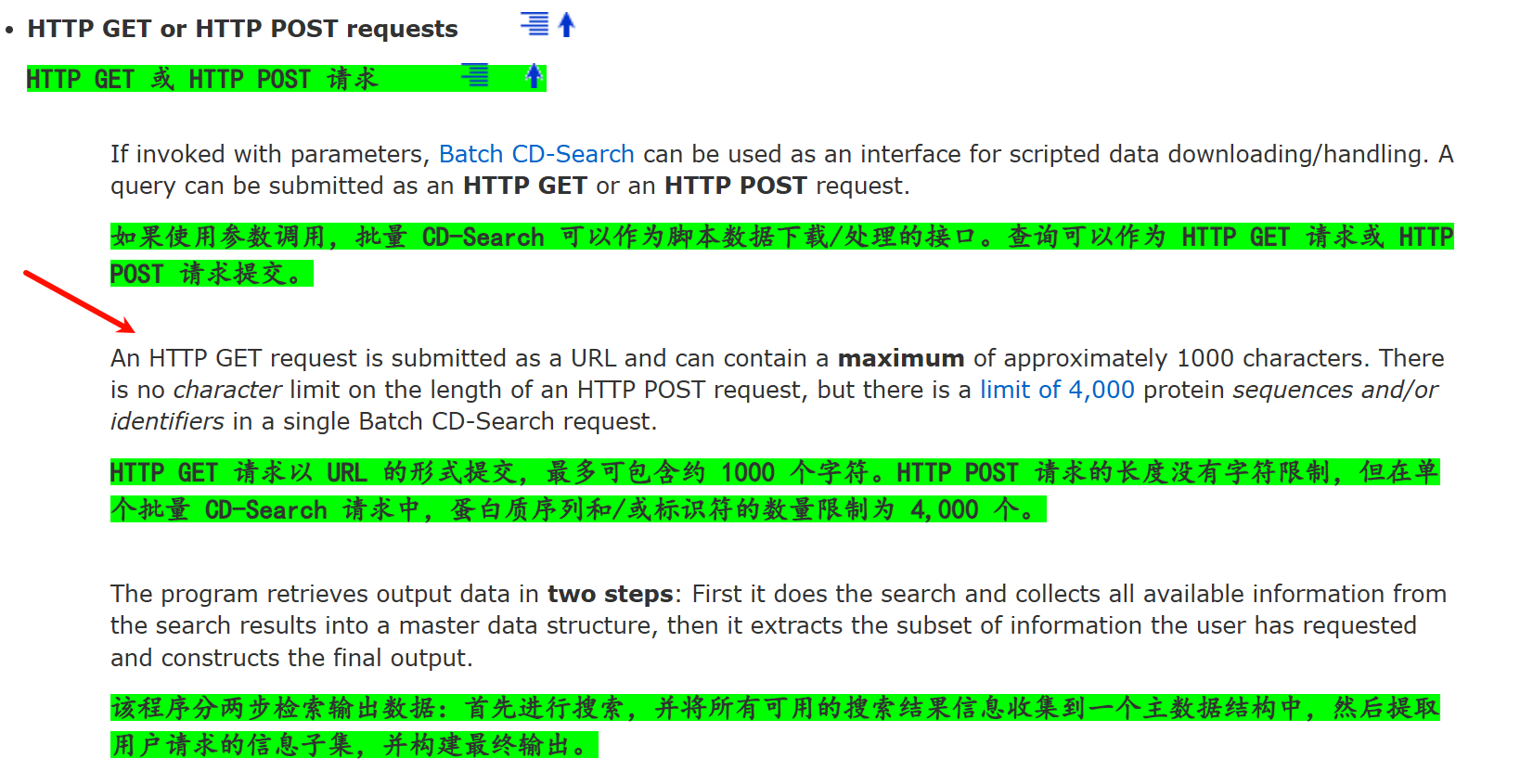
其中参数设置方面参考:
https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml#BatchRPSBWebAPI

具体解释参考:https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml#TOC_BatchCDSearch
官方提供了一个Perl脚本实例,来进行数据申请分析:



plain
#!/usr/bin/perl -w
use strict;
use LWP::UserAgent;
use Getopt::Std;
###############################################################################
# URL to the Batch CD-Search server
###############################################################################
my $bwrpsb = "/Structure/bwrpsb/bwrpsb.cgi";
###############################################################################
# read list of queries and parameters supplied; queries specified in list piped
# from stdin
###############################################################################
my @queries = <STDIN>;
my $havequery = 0;
###############################################################################
# do some sort of validation and exit if only invalid lines found
###############################################################################
foreach my $line (@queries) {
chomp($line);
if ($line =~ /[a-zA-Z0-9_]+/) {
$havequery = 1;
}
}
if ($havequery == 0) {
die "No valid queries!\n";
}
###############################################################################
# set default values
###############################################################################
my $cdsid = "";
my $cddefl = "false";
my $qdefl = "false";
my $smode = "auto";
my $useid1 = "true";
my $maxhit = 250;
my $filter = "true";
my $db = "cdd";
my $evalue = 0.01;
my $dmode = "rep";
my $clonly = "false";
my $tdata = "hits";
###############################################################################
# deal with command line parameters, change default settings if necessary
###############################################################################
our($opt_d, $opt_e, $opt_F, $opt_b, $opt_t, $opt_s, $opt_a, $opt_q);
getopts('d:e:F:b:t:s:a:q');
if ($opt_d) {
$db = $opt_d;
print "Database option set to: $db\n";
}
if ($opt_e) {
$evalue = $opt_e;
print "Evalue option set to: $evalue\n";
}
if ($opt_F) {
if ($opt_F eq "F") {
$filter = "false"
} else {
$filter = "true";
}
print "Filter option set to: $filter\n";
}
if ($opt_b) {
$maxhit = $opt_b;
print "Maxhit option set to: $maxhit\n";
}
if ($opt_t) {
$tdata = $opt_t;
print "Target data option set to: $tdata\n";
}
if ($opt_s) {
$clonly = "true";
print "Superfamilies only will be reported\n";
}
if ($opt_a) {
$dmode = "all";
print "All hits will be reported\n";
}
if ($opt_q) {
$qdefl = "true";
print "Query deflines will be reported\n";
}
###############################################################################
# submitting the search
###############################################################################
my $rid;
{
my $browser = LWP::UserAgent->new;
my $response = $browser->post(
$bwrpsb, Content_Type => 'multipart/form-data',
Content => [
'useid1' => $useid1,
'maxhit' => $maxhit,
'filter' => $filter,
'db' => $db,
'evalue' => $evalue,
'cddefl' => $cddefl,
'qdefl' => $qdefl,
'dmode' => $dmode,
'clonly' => $clonly,
'tdata' => "hits",
( map {; queries => $_ } @queries )
],
);
die "Error: ", $response->status_line
unless $response->is_success;
if($response->content =~ /^#cdsid\s+([a-zA-Z0-9-]+)/m) {
$rid =$1;
print "Search with Request-ID $rid started.\n";
} else {
die "Submitting the search failed,\n can't make sense of response: $response->content\n";
}
}
###############################################################################
# checking for completion, wait 5 seconds between checks
###############################################################################
$|++;
my $done = 0;
my $status = -1;
while ($done == 0) {
sleep(5);
my $browser = LWP::UserAgent->new;
my $response = $browser->post(
$bwrpsb,
[
'tdata' => "hits",
'cdsid' => $rid
],
);
die "Error: ", $response->status_line
unless $response->is_success;
if ($response->content =~ /^#status\s+([\d])/m) {
$status = $1;
if ($status == 0) {
$done = 1;
print "Search has been completed, retrieving results ..\n";
} elsif ($status == 3) {
print ".";
} elsif ($status == 1) {
die "Invalid request ID\n";
} elsif ($status == 2) {
die "Invalid input - missing query information or search ID\n";
} elsif ($status == 4) {
die "Queue Manager Service error\n";
} elsif ($status == 5) {
die "Data corrupted or no longer available\n";
}
} else {
die "Checking search status failed,\ncan't make sense of response: $response->content\n";
}
}
print "===============================================================================\n\n";
###############################################################################
# retrieve and display results
###############################################################################
{
my $browser = LWP::UserAgent->new;
my $response = $browser->post(
$bwrpsb,
[
'tdata' => $tdata,
'cddefl' => $cddefl,
'qdefl' => $qdefl,
'dmode' => $dmode,
'clonly' => $clonly,
'cdsid' => $rid
],
);
die "Error: ", $response->status_line
unless $response->is_success;
print $response->content,"\n";
}上述bwrpsb.pl脚本,直接在终端中运行
plain
./bwrpsb.pl < samplefile.in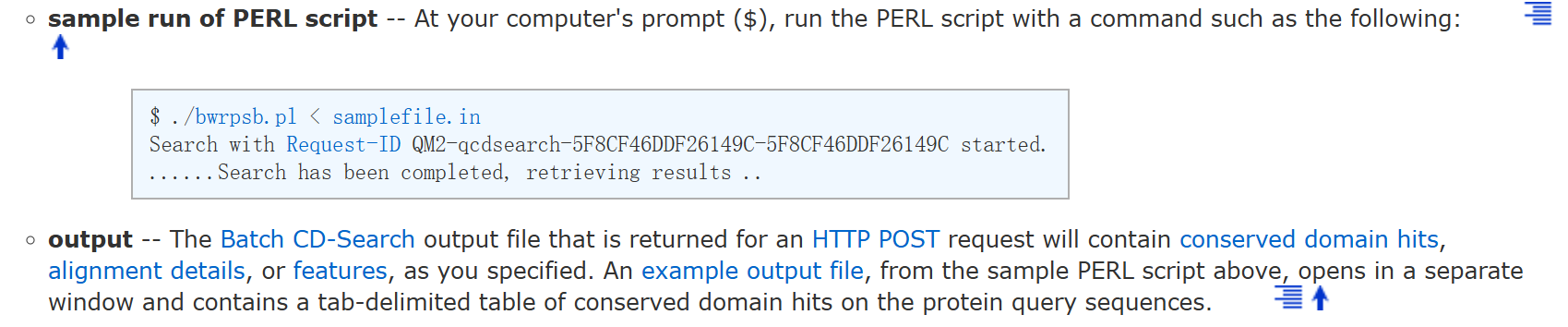
能够获取数据,但是没法整合到目前项目的python pipeline中。
无法简单调用api接口,对于任意一条序列进行call domain,虽然能够获取数据,但是更想把数据获取方法整合到流程中,而不是单独一个py或者pl单独处理一个大型文件就能够直接完成,个人是更倾向于在整体流程中用循环来处理批量数据,对于单个序列注释的domain能够做一点中间其他的分析。
可以参考:https://github.com/qeubic/protein_features
https://github.com/xpolak37/output_merger
3,结构域绘制:结构域可视化/结构域绘制
以边界和蛋白质长度为输入,绘制蛋白质结构域
(1)PROSITE结构域数据库server:
https://prosite.expasy.org/mydomains/
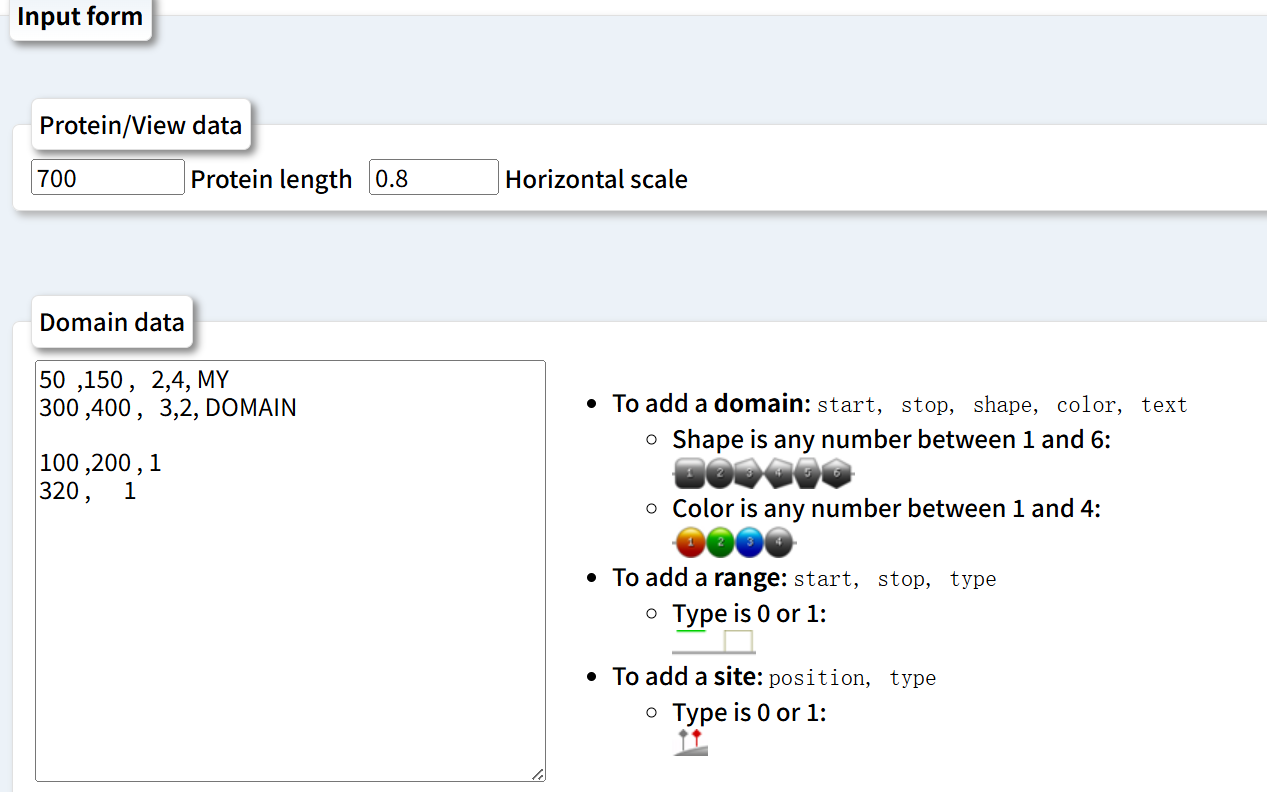

(2)Perl脚本大户DomainDraw
https://github.com/doktor-nick/DomainDraw
参考:https://www.biostars.org/p/11187/
TED:
https://ted.cathdb.infohttps://www.cathdb.info/
总结:
个人不是很喜欢CDD数据库,印象中NCBI系的可视化界面都挤在一起,而且基本上蛋白质数据库很少使用NCBI系,印象中基本上都使用EMBL-EBI欧洲那边数据库多点,
没怎么在文献中看到有用这个数据库的,接口也不是我喜欢的,虽然能够获取数据。
3,HAMAP(https://hamap.expasy.org/)

也是一个蛋白质注释数据库





重点:如何使用&接口访问
(1)单个序列
使用HAMAP-Scan,注意这是一个web interface,也就是网页版在线工具;
单个序列的话,直接输入
(2)多个序列:使用HAMAP-Scan
参考https://hamap.expasy.org/hamap-scan/hamap_scan_userman.html
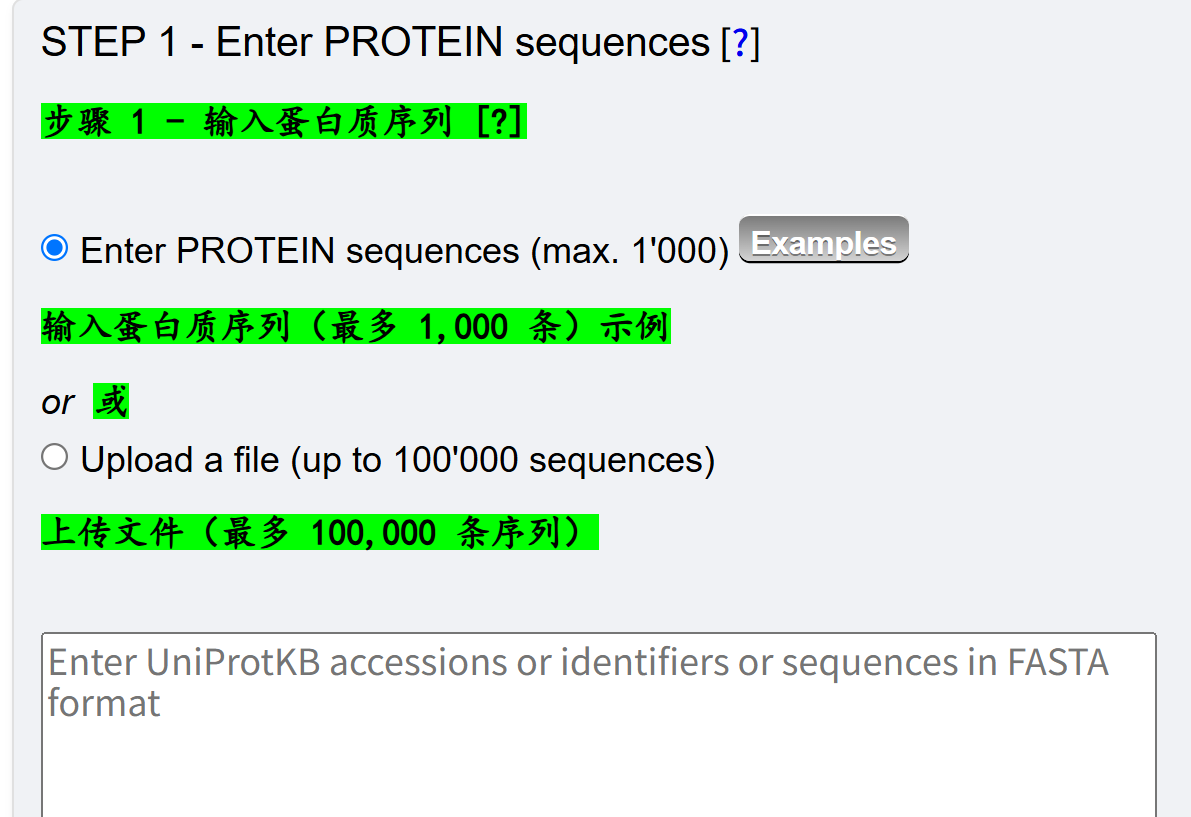
多个序列的话,直接上传fasta文件,
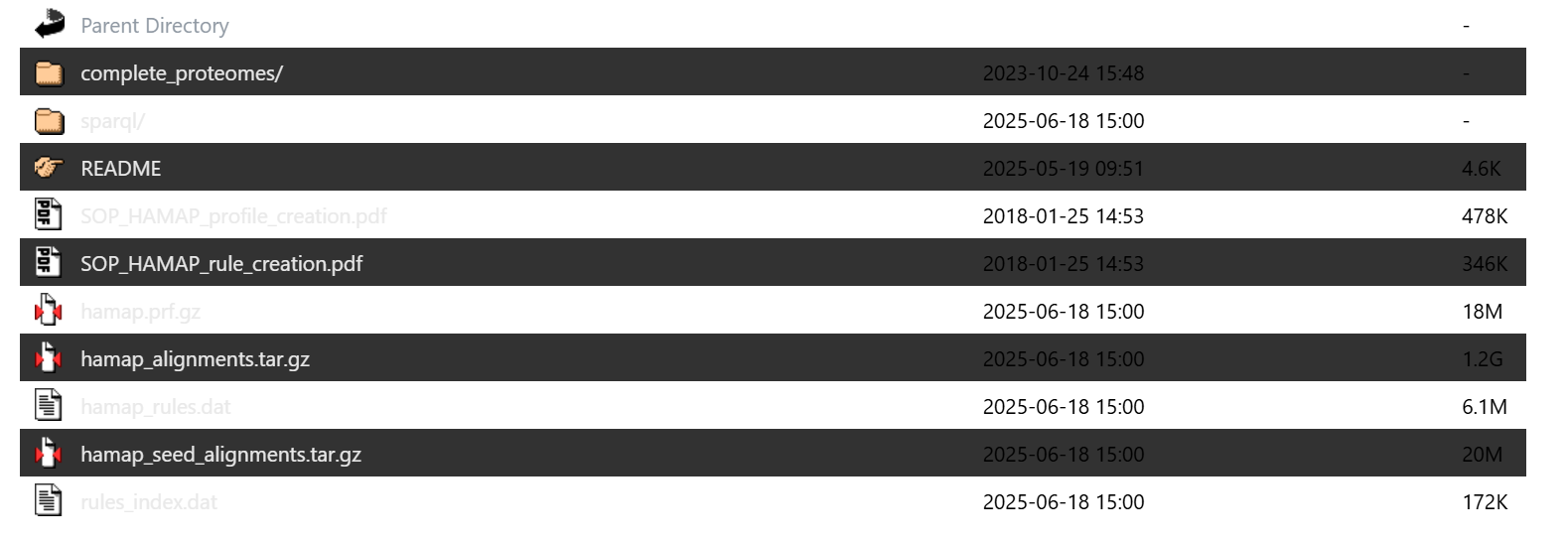
其中注释规则、配置文件和比对结果,可以从https://ftp.expasy.org/databases/hamap/中下载,
一旦工作提交到服务器上之后,后续就是等待的时间,
比如说我提交了这么一个数据集,

可以看到,暂时没有找到api接口,可以从中干预或者是整合到python 流程中,
还是鸡肋的输入批量文件+直接等结果,不能够个性化分析。
下面是我的测试结果,可以看到800条序列接近1k条序列,大概30mins左右就能够完成分析;

但是分析结果一言难尽:


总结:
暂时没看到api接口,更别提流程整合了,虽然操作很傻瓜式,但是不能个性化分析,不爽,暂时不是一个合格的数据来源。
4,NCBIFAM(https://www.ncbi.nlm.nih.gov/genome/annotation_prok/evidence/)


看起来更像是蛋白质注释数据库(蛋白质名称id分配+属性分配),而不是单一的结构域注释,

主要使用3种类型的数据来进行辅助注释:
Hidden Markov Models (HMMs), BlastRules, and domain architectures.



1. 隐马尔可夫模型(HMMs)
- 是什么?
HMMs 是一种数学模型,用来判断一个蛋白质是否属于某个特定的蛋白质家族。简单来说,它就像是一个"家族鉴定器"。 - 怎么用?
研究人员会收集已知功能的蛋白质序列,然后通过复杂的计算生成一个模型(HMM)。当一个新的蛋白质序列出现时,就可以用这个模型来判断它是否属于某个家族。如果匹配度很高,就可以给这个新蛋白质一个名字和相关的功能描述。 - 例子
比如有一个蛋白质家族是"阳离子转运蛋白家族"。科学家们通过 HMM 模型,可以判断一个新的蛋白质是否属于这个家族,并给它命名为"阳离子转运蛋白"。
关于更一般的HMM的一些简单拓展,可以参考之前的博客:
https://blog.csdn.net/weixin_62528784/article/details/150872038?spm=1001.2014.3001.5502
2. BlastRules
- 是什么?
BlastRules 是一种基于序列比对的规则。它通过比较蛋白质序列之间的相似性来判断功能。 - 怎么用?
同样挑选一些已知功能的"模型蛋白质",然后设定一些比对的阈值(比如序列相似度和覆盖率)。如果一个新的蛋白质和这些模型蛋白质在序列上非常相似,就可以认为它具有类似的生物学功能。 - 例子
比如有一个模型蛋白质是"锌外排转运蛋白",如果一个新的蛋白质在序列上和它非常相似(比如相似度超过 90%),就可以把它命名为"锌外排转运蛋白"。
3. 结构域架构
- 是什么?
结构域架构是指蛋白质中保守的结构域(功能区域)的排列顺序。蛋白质可以由多个结构域组成,这些结构域的组合方式可以帮助判断蛋白质的功能。 - 怎么用?
根据已知的结构域组合来分类蛋白质。如果一个新的蛋白质有特定的结构域组合,就可以根据这个组合来推断它的功能。 - 例子
比如一个蛋白质包含两个特定的结构域 A 和 B,而这种组合通常与"信号传导功能"相关,那么这个新蛋白质就可以被命名为"信号传导蛋白"。
总的来说,我们大概能够理解这个数据库构建的一些原理。
但是我暂时没有找到这个数据库的api接口,或者是能够利用上这个数据库的interface。
5,PANTHER(https://pantherdb.org/)


更像是一个蛋白质分类数据库(更加侧重于蛋白质进化关系,可能结构、结构域只是辅助分类的一部分信息)


同样是人工编辑+算法预测收集:
 至于序列分类,其实和结构域注释,都可以用于蛋白质识别。
至于序列分类,其实和结构域注释,都可以用于蛋白质识别。
重点:如何使用&接口访问
我们先试一下下面的这个"sequence search":

同样的,我还是以CTCF human为例,只输入其氨基酸序列:
可以发现,基本上都是先识别该序列(所以有一个hit),其实并非结构域注释,顶多在确定"有名有姓"之后再援引蛋白质注释数据库。
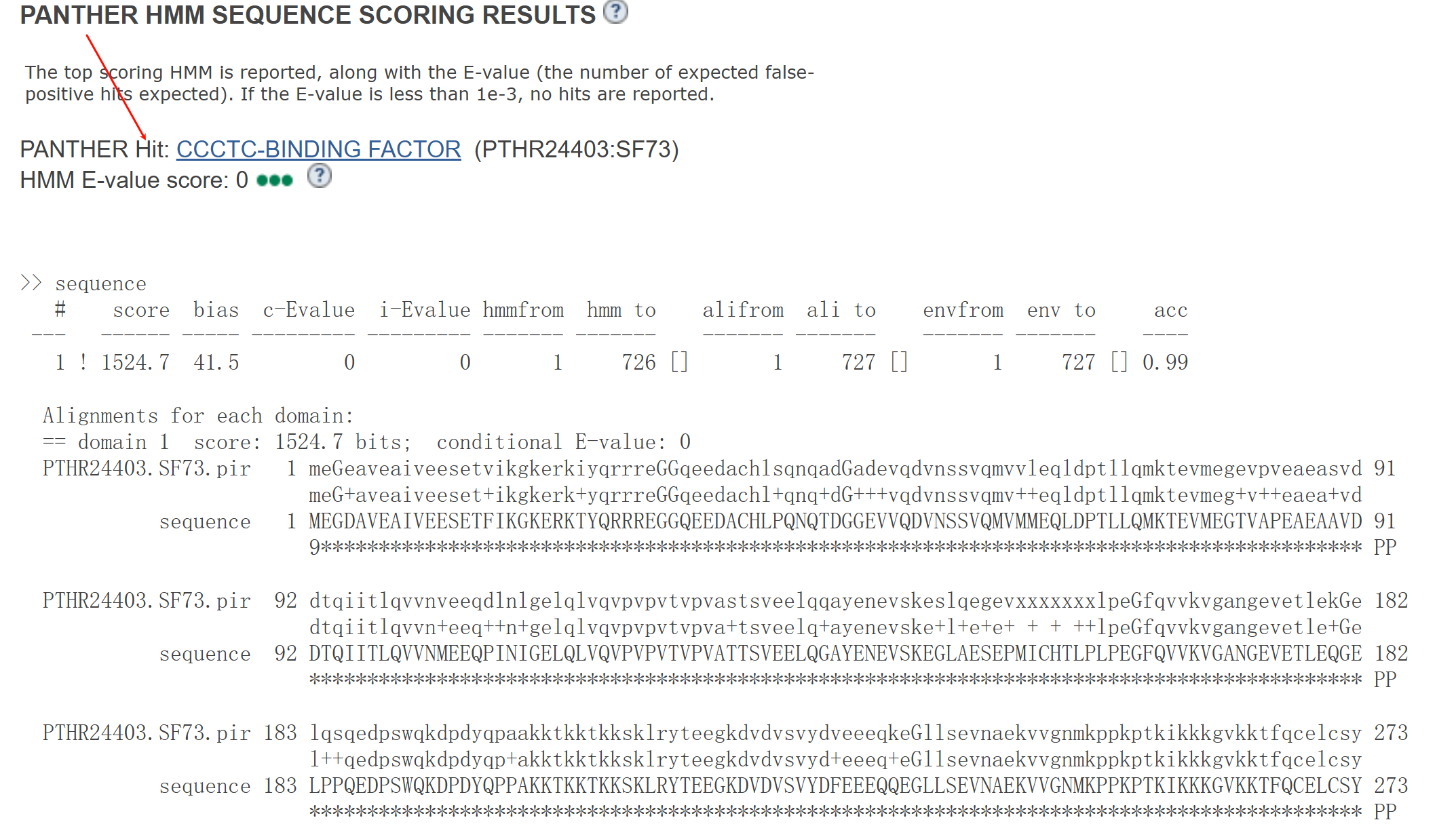
并没有达到从头注释序列结构域的目的,pass。
还有一个是对gene list做的分析,不符合我们的目的,pass。
总结:
暂时没有找到API接口,这个数据库其实更侧重于与gene数据的联动,个人用的比较少。
6,PIRSF(https://proteininformationresource.org/pirsf/)


分类的基础,包含结构域注释。
对于PIR和其他数据库的关系,比如说Uniprot:


重点:如何使用&接口访问
参考界面:https://proteininformationresource.org/pirwww/search/
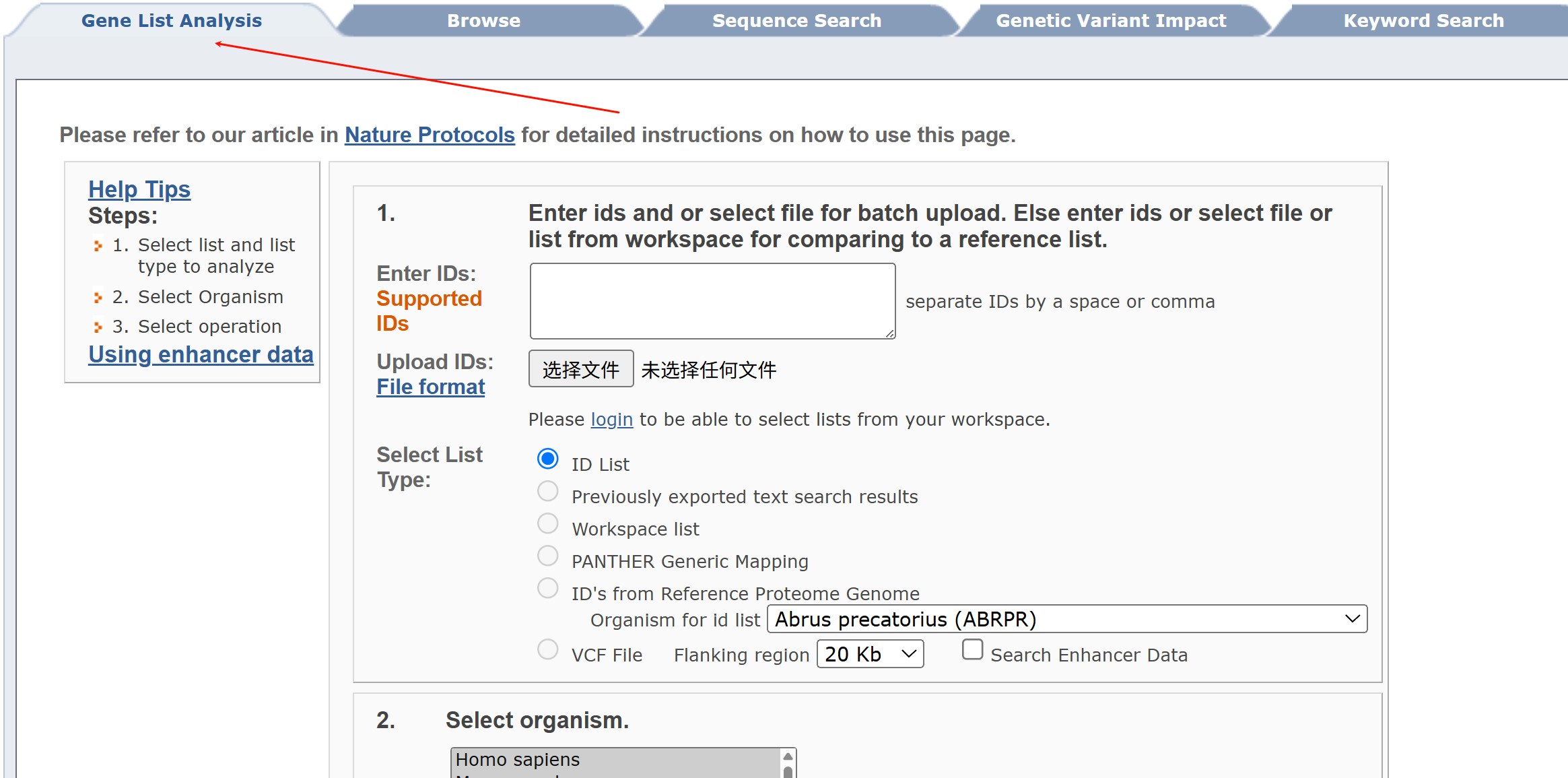
比如说下面的"search & analysis tools",
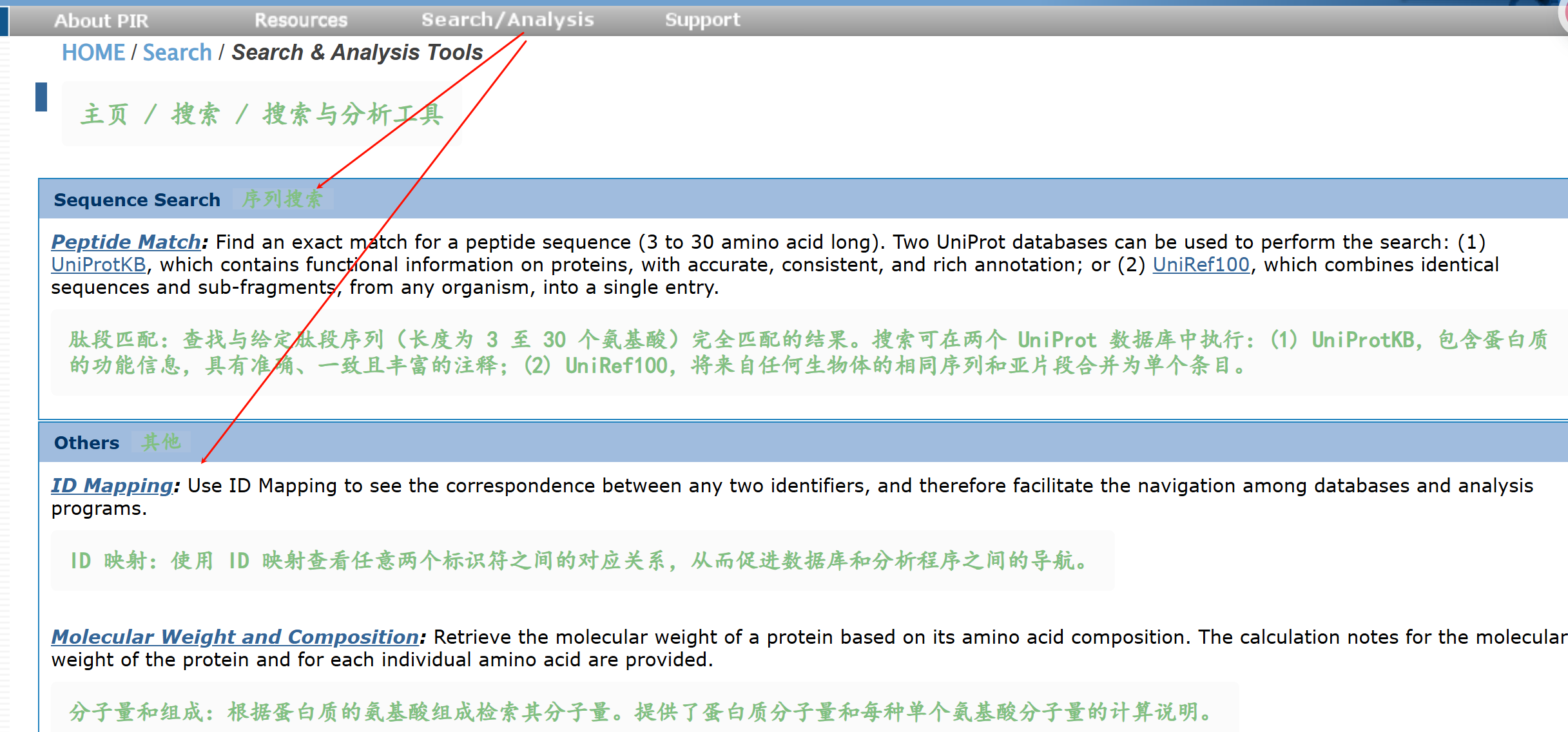
其实更像是蛋白质id注释,序列匹配(Blast那一套),基本上和前面的思路没什么区别,都是先序列匹配之后,获取hit也就是id注释,再对id进行结构域注释数据库引用,但其本身并没有做结构域注释。
另外主页面上也有搜索链接:
Batch链接见:https://proteininformationresource.org/pirwww/search/batch_sf.shtml
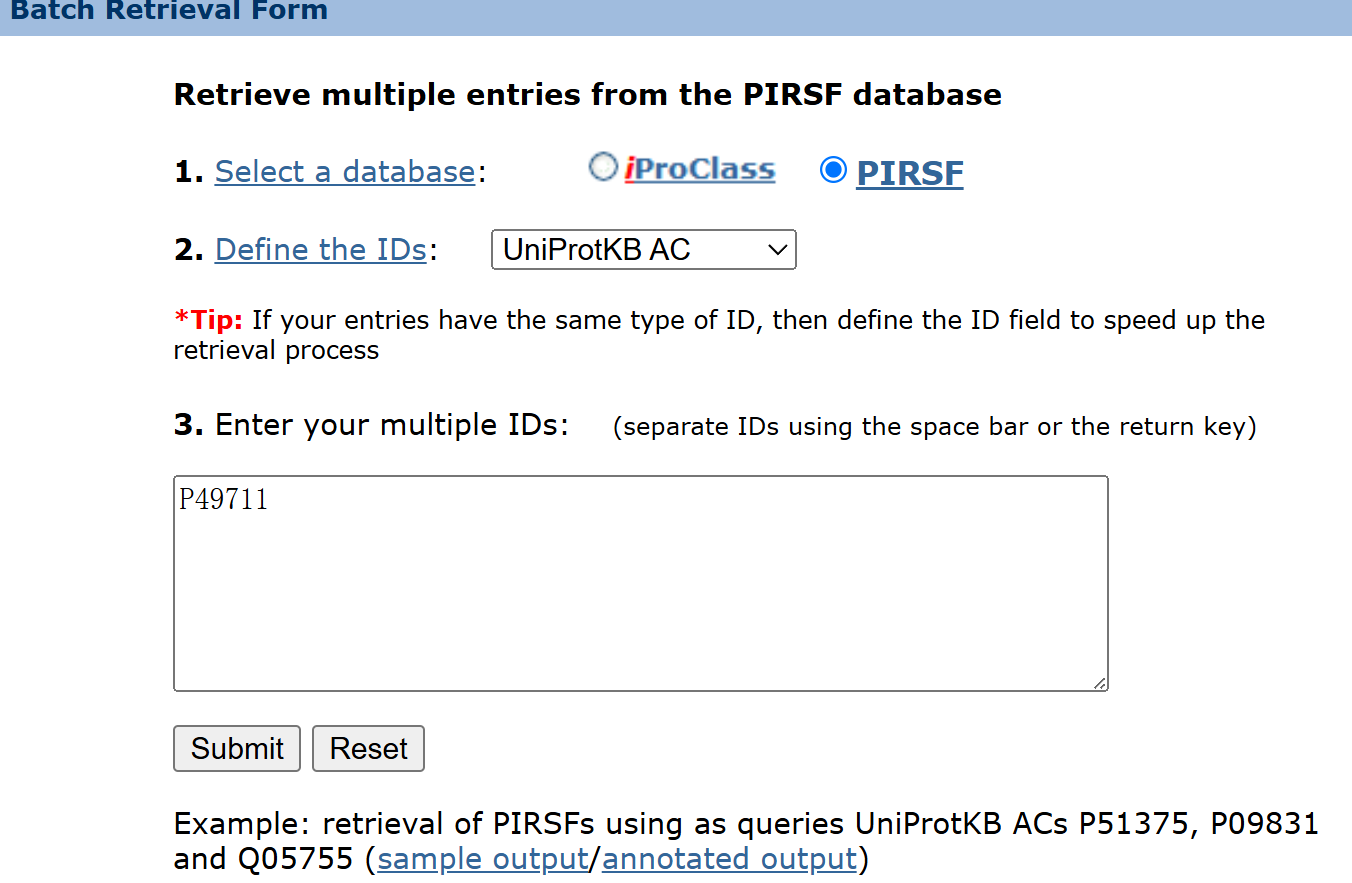
但是实测无法正常使用:
下面那个predict的链接,见:https://research.bioinformatics.udel.edu/PIRSitePredict/

也是直接无法使用,感觉都像是很久没有经过维护。
总结:
API接口并没有找到,一些web server甚至也不能正常访问,不过好在这部分数据如果是整合到Uniprot中的话,其实就是前面(一)中尝试的内容。
7,PRINTS(https://interpro-documentation.readthedocs.io/en/latest/prints.html)

可以看得出来,是存储蛋白质分类所需的motif数据,类似于HMM profile,PSSM之类,
存储分类、注释的特征数据。

上面这里的最后一行我们可以看到,该数据库已经停用了,已经归档在InterPro中,类似于Github中归档的仓库,已经没有人再持续维护更新了,
包括这个标题里的链接,其实只能传送到InterPro文档指南,确实没有实际的数据库Url,所以确实是停止更新了。
所以直接pass,但是这个fingerprints和motifs的关系倒是挺有意思的。
8,Pfam(https://pfam.xfam.org/)
新址:https://www.ebi.ac.uk/interpro/entry/pfam/#table

这个在我印象中,看到文献里用的其实比较多,包括一些深度学习序列分析,在数据集处理的时候。
从上面最后一行,我们可以看到,这个数据库Pfam已经归并到InterPro中了。
我在本节标题中给出的链接是旧址: https://pfam.xfam.org/

其实看被引用的文献:
还在2021年,这是一个好消息,5年之内,至少这个数据库的时效性还是有一定参考价值的。

在旧址上,我还是以CTCF为例(此处输入Uniprot的ID号),

跳转界面如下:
https://www.ebi.ac.uk/interpro/search/text/P49711/#table
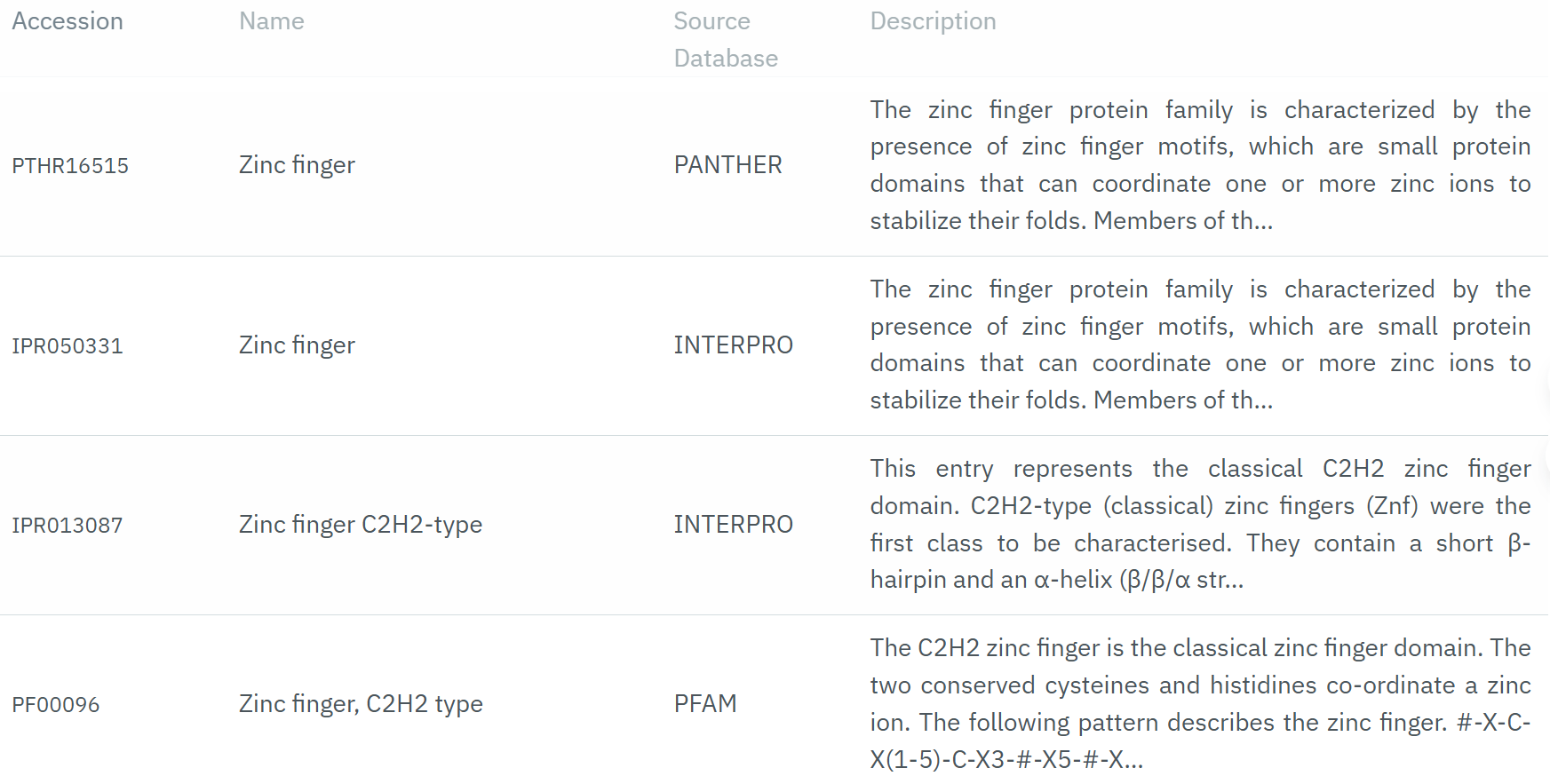
分别是各个成员数据库的hit记录,
我们以上面的这个Pfam数据库hit为例,因为我们这一节要看的就是Pfam数据库:
界面链接参考:https://www.ebi.ac.uk/interpro/entry/pfam/PF00096/
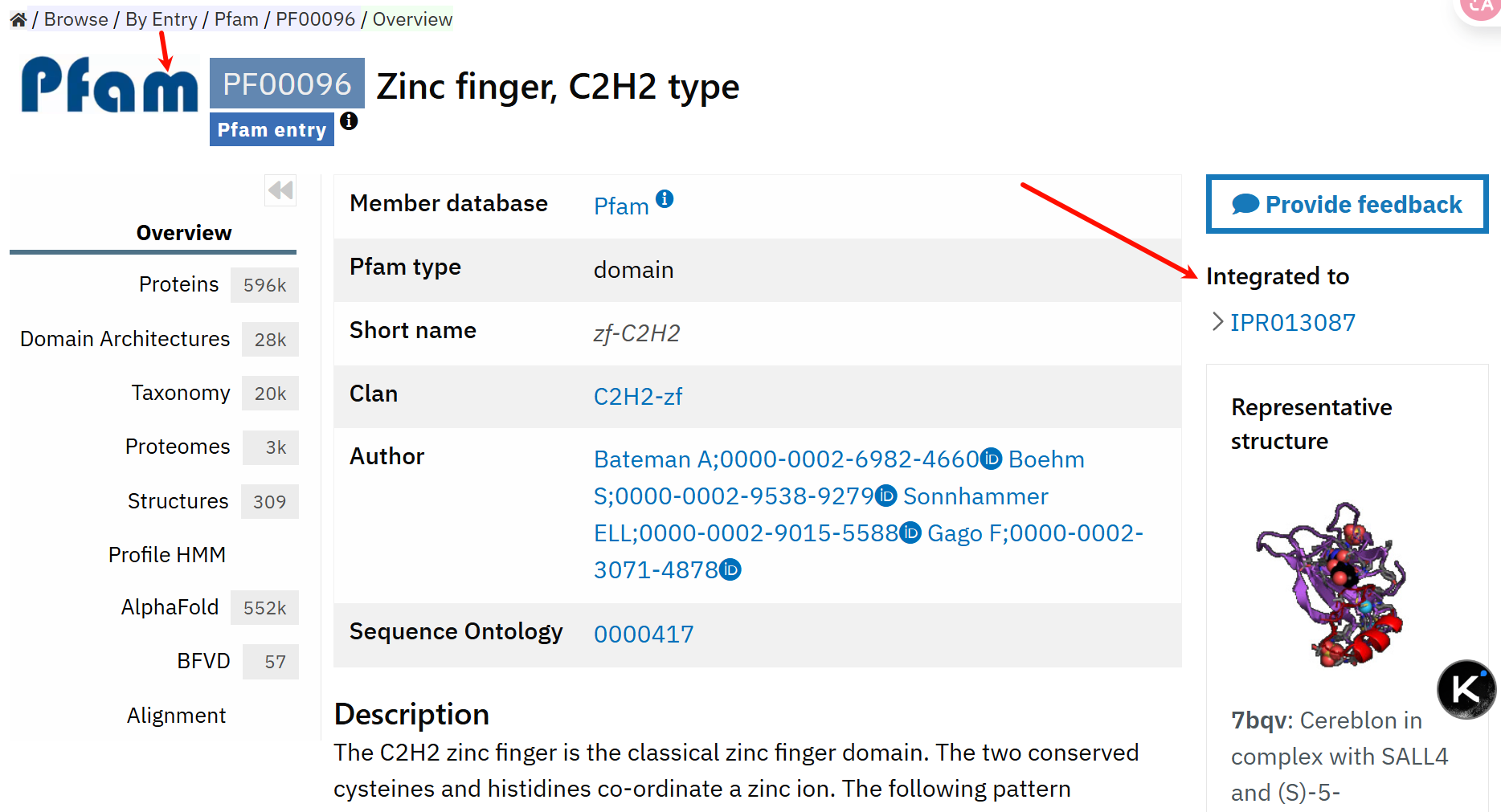
我们可以看到,这个其实就可以算是C2H2 Zinc Finger的domain注释了,
就是在Pfam数据库中,检索到了C2H2 ZF的domain特征谱的hit。


另外新址:https://www.ebi.ac.uk/interpro/entry/pfam/#table
总结:
在旧址中没有找到API接口,但是因为整合到InterPro中了,所以其实可以调用后者的API来解决问题,后者的API放后面讲,此处忽略。
9,PROSITE profiles/PROSITE patterns(https://prosite.expasy.org/)


这两个数据库,其实Url链接是一致的,都是指向PROSITE数据库。

总的来说,其实和前面数据库大差不差,前面有的数据库存fingerPrints,有的存motif,
有的存多序列比对MSA文件、以及HMM谱,
这个数据库存储的是广义domain(功能性位点)的一些特征谱,比如说如何定义的rules(其实涉及到rules,就是一个好消息了,表明这个是底层原理部分,我们是可以直接用于序列分析的,而不是调用其他人数据库的二手资料)。

就是引用文献有点老了:

关于Prorule,参考链接:https://prosite.expasy.org/prorule_details.html


重点:如何使用&接口访问
我们先看一下该数据库中所提供的web server:
链接见:https://prosite.expasy.org/scanprosite/
此处我还是以CTCF为例子,以fasta格式仅输入序列数据(id是我自己编的)
当然也可以输入其他格式数据,输出结果如下:
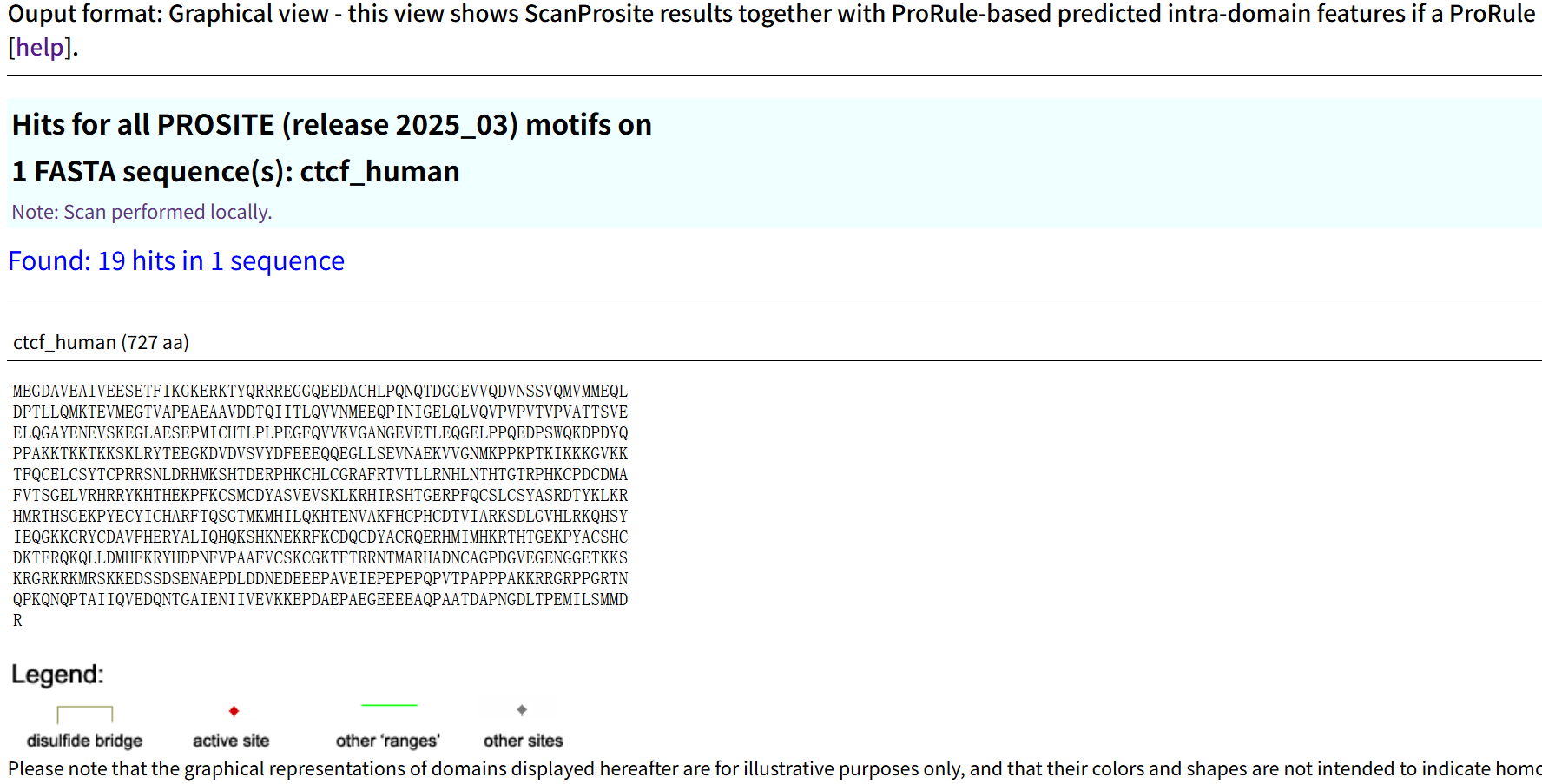
我们可以看到有19 hits,而Zn Finger是11 hits(符合前面的结论):
首先是11个PS50157 profile的hit,其实就是Zn Finger

具体PS50157规则的细节,链接参考:https://prosite.expasy.org/cgi-bin/prosite/nicedoc.pl?PS50157


具体的生物学背景:描述、起源、以及发现历史,援引文献都有,暂不赘述,pass。

一些计算手段与真实匹配检测的注意点:

当然,我们再仔细查看一下:
除了这个界面中所hit到的PS50157结构域轮廓(matrix rule),

其实还有另外一个规则rule,也就是下面的PS00028

所以这里有一个问题:对于同一个domain,其实Prosite数据库中也有不同的Rule来call识别,
并且可能hit注释的数据也有可能不一样:
这里,我们再次回到最前面Uniprot数据库中CTCF的注释:
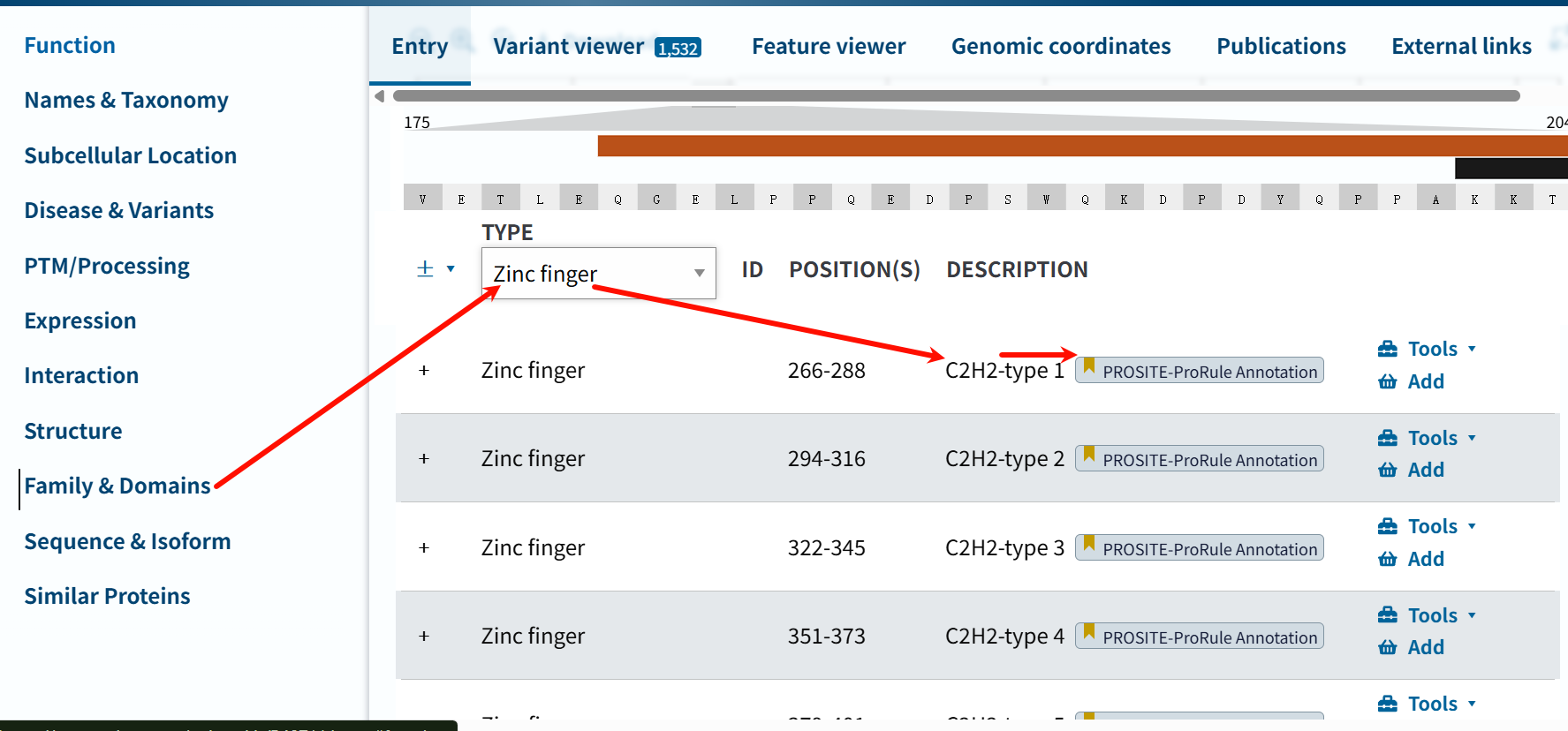
每一个注释我都点进去看了,指向的其实并不是简单的matrix或者是pattern,而是ProRule文件,链接见https://prosite.expasy.org/unirule/PRU00042

可以看的出来,这里用的就是Prosite注释规则(或者说用了Prosite数据库),来注释锌指结构域,问题就是为什么没有使用其他数据库?
我们再仔细比对一下数据:
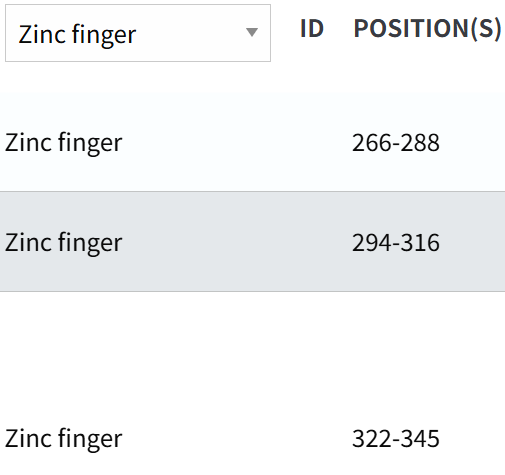
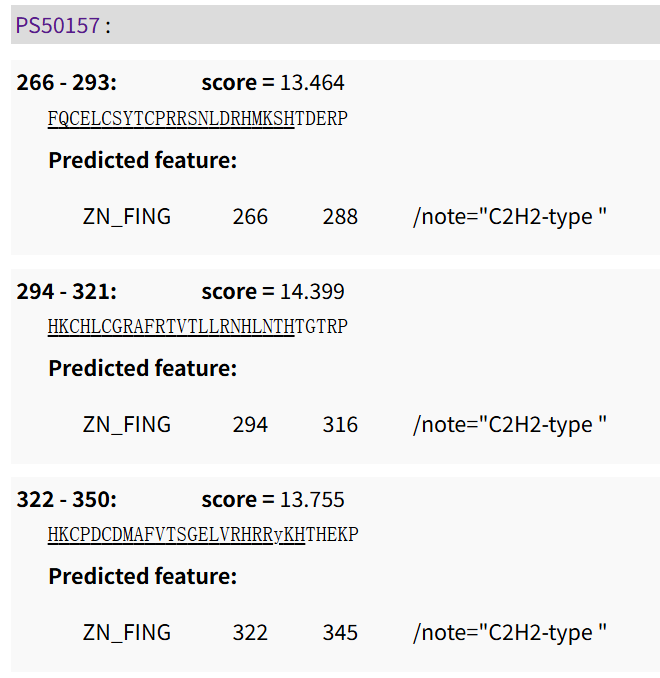
确定了是一样的坐标,就是注释数据的来源。
另外的8个hits,是pattern,也是C2H2 ZF的,但是数目比matrix识别出来的少,
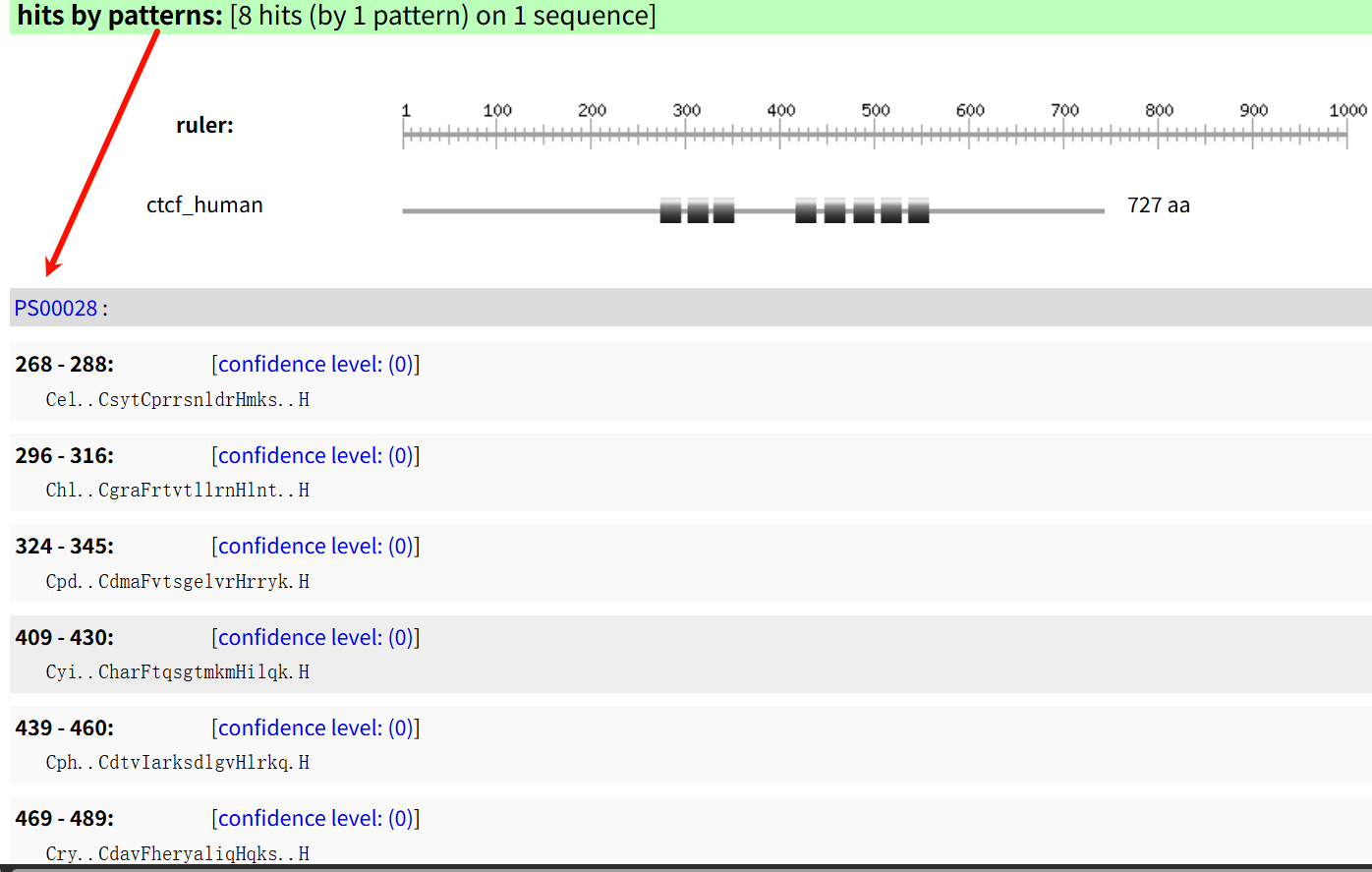
而且我们可以看到就算是识别出来的,在坐标数据上也不一定对应:
这个是matrix识别出来的第2个ZF
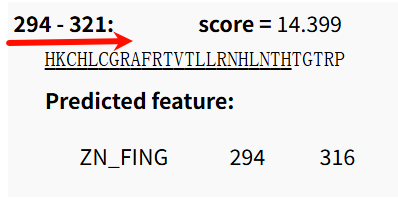
这个是pattern识别出来的第2个ZF
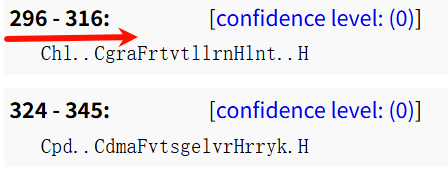
我们可以看到数据上是有区别的,坐标index不同、数目也不同,一个识别出来了11个,另外一个只识别出来了8个。
现在再次回到我们最前面CTCF蛋白的Uniprot数据库注释部分:
https://www.uniprot.org/uniprotkb/P49711/entry#family_and_domains

确实就能够理解这里的(11,8)hits数据了。
输出也有很多格式,具体可以参考:
https://prosite.expasy.org/scanprosite/scanprosite_doc.html#of_graphical
总结:
暂时没有找到API接口,但是鉴于这个数据库也是整合在InterPro中,同理Pfam,我们可以放到InterPro的接口中去叙述。
题外话:关于PROSITE数据库的探究,可以参考之前的博客:
10,SFLD(http://sfld.rbvi.ucsf.edu/archive/django/index.html)
新址:https://www.ebi.ac.uk/interpro/entry/sfld/#table



这也是一个旧址,
参考:https://interpro-documentation.readthedocs.io/en/latest/sfld.html

该数据也是归档到InterPro中,并不再维护更新了,旧数据倒是还可以通过InterPro访问。
11,SMART(https://smart.embl.de/)

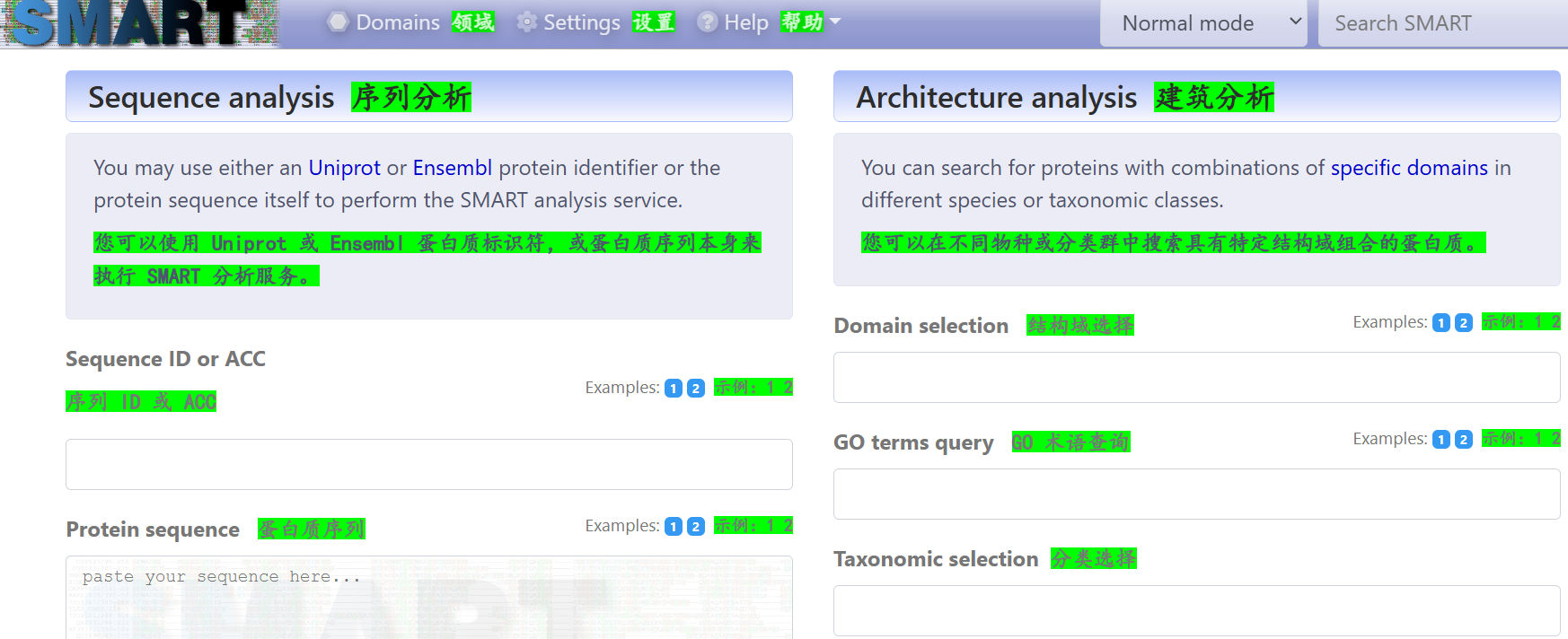
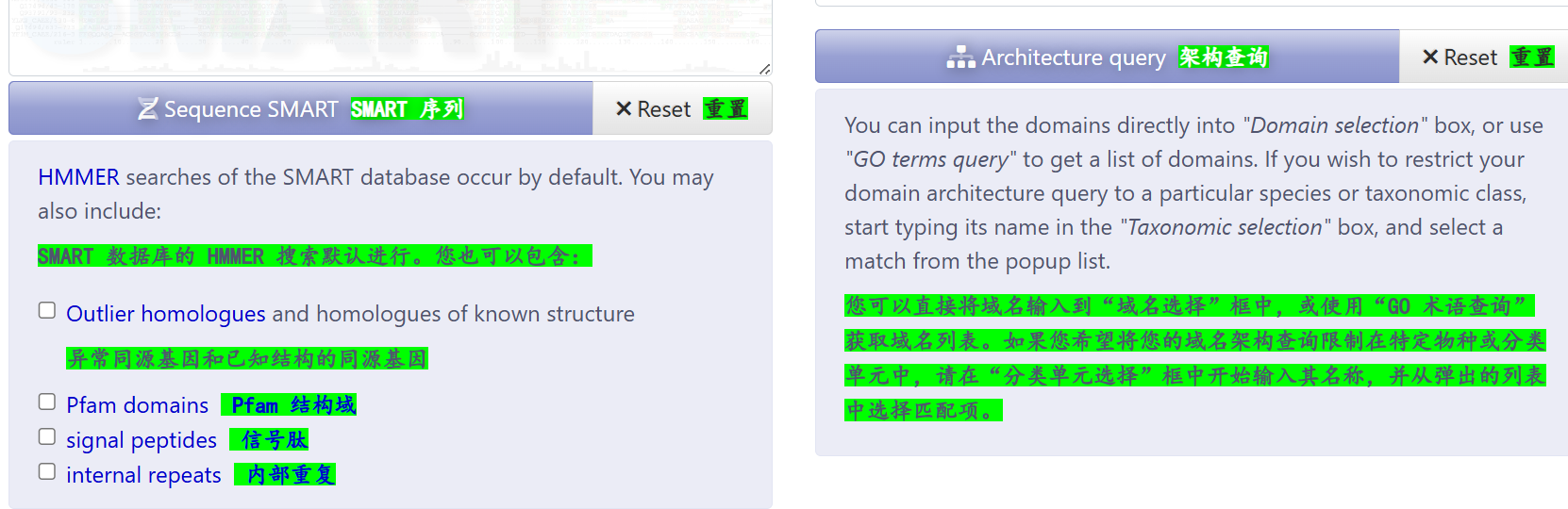
重点:如何使用&接口访问
序列分析:同样的我还是以CTCF为例,仅输入序列数据,并没有输入指定"已知有名有姓"蛋白质的其他数据库ID,
链接参考:https://smart.embl.de/smart/show_motifs.pl
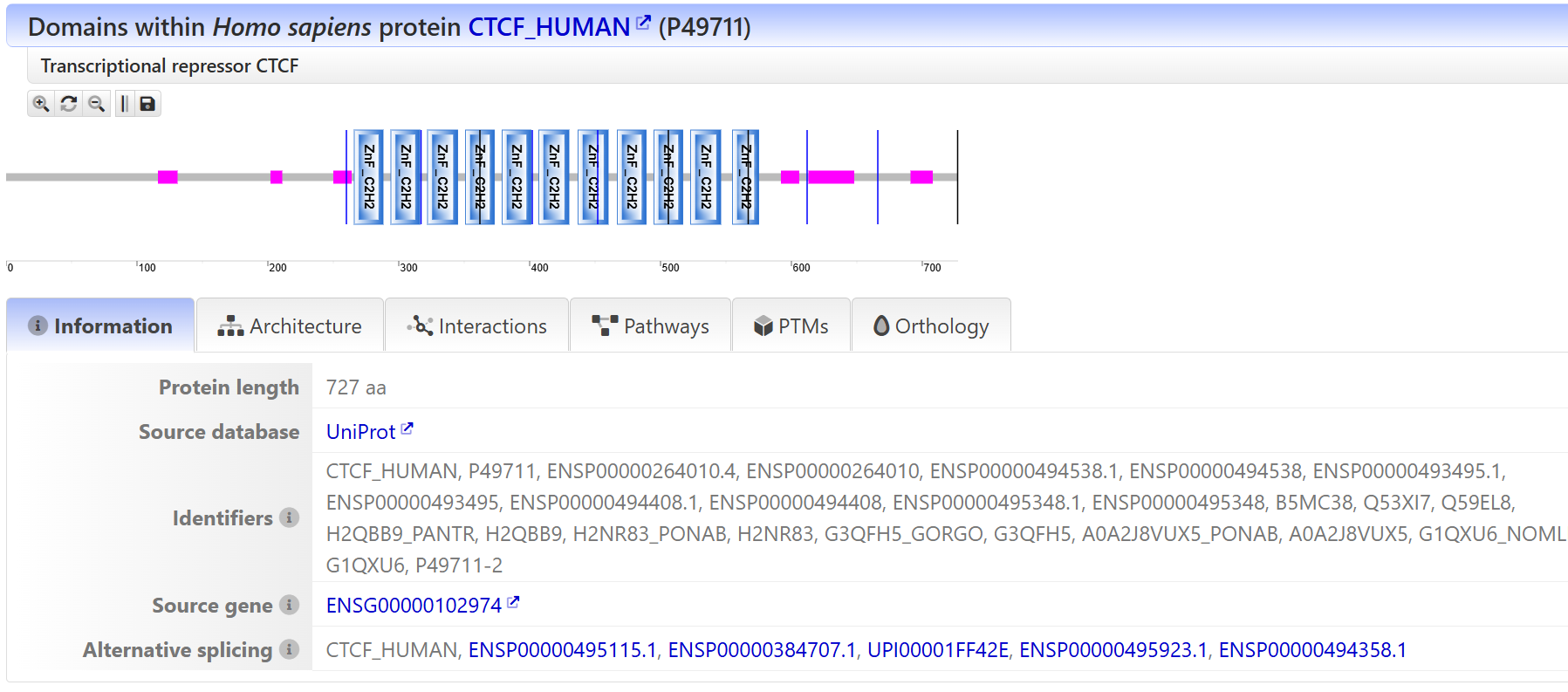
从上面结果可以看到,搜索结果其实还是先识别,先识别出极有可能是什么蛋白(这里就是CTCF human,给出了Uniprot数据库中有名有姓的ID);
可以看到,结果中给出了11个ZF 的hit,是和前面部分数据库预测结果吻合的;
除了ZF锌指,还给出了低复杂度区域的hit,
此处的低复杂度区域,其实注释倒是出我意外,因为低复杂区域和disorder注释其实还有有点区别的,
我知道Uniprot中的disorder无序区域其实使用了MobiDB数据库的注释,
对于低复杂度区域其实少注释的,但SMART数据库能够给出这个注释。
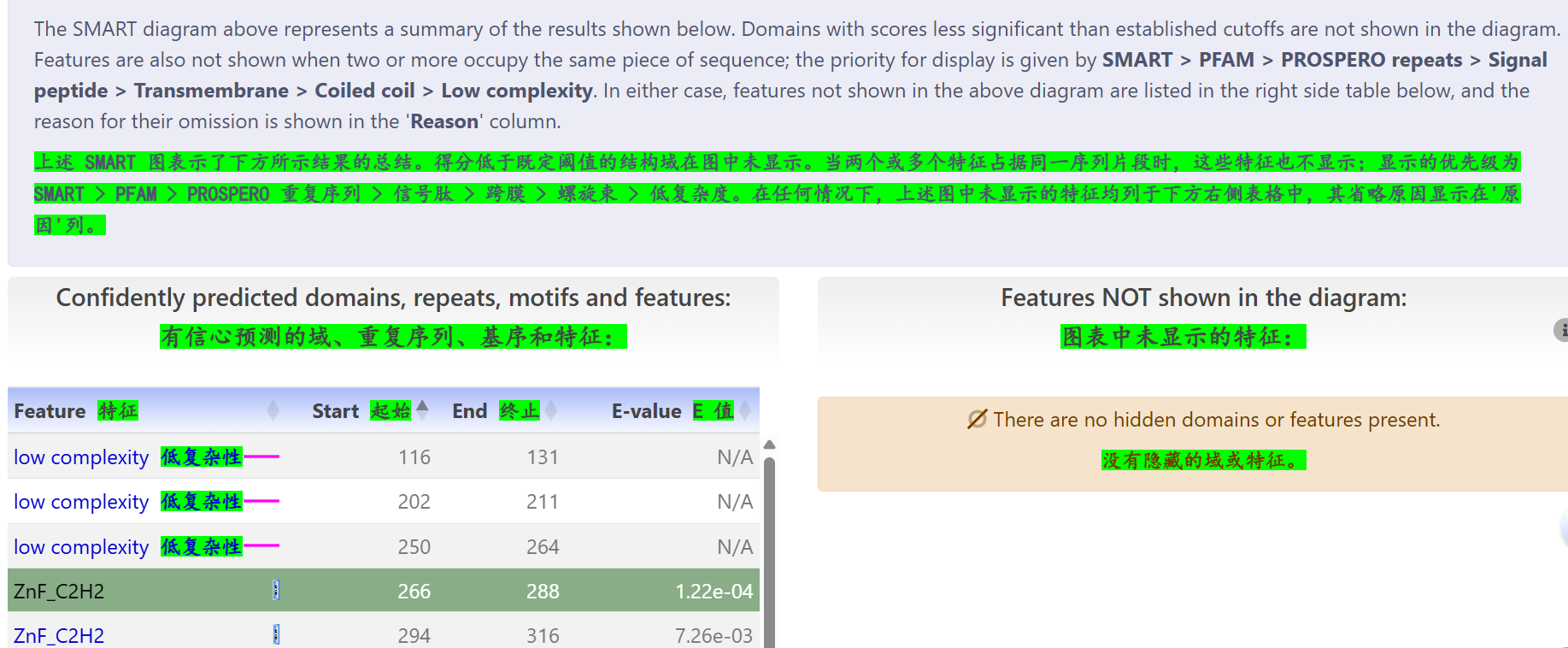
刚才我们看的只是"information"部分,其实还可以看一看其他的部分
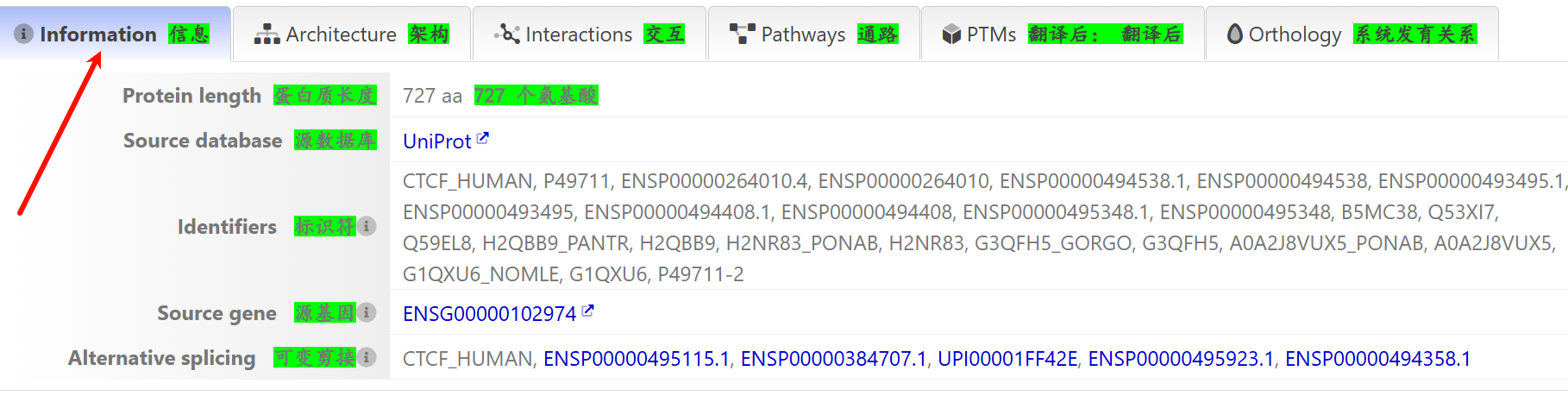
结构域架构分析:
这个功能其实很好,可以从结构域的排列组合,也就是pattern的角度去分析进化、亲缘关系,然后获取其他相关的蛋白质数据。
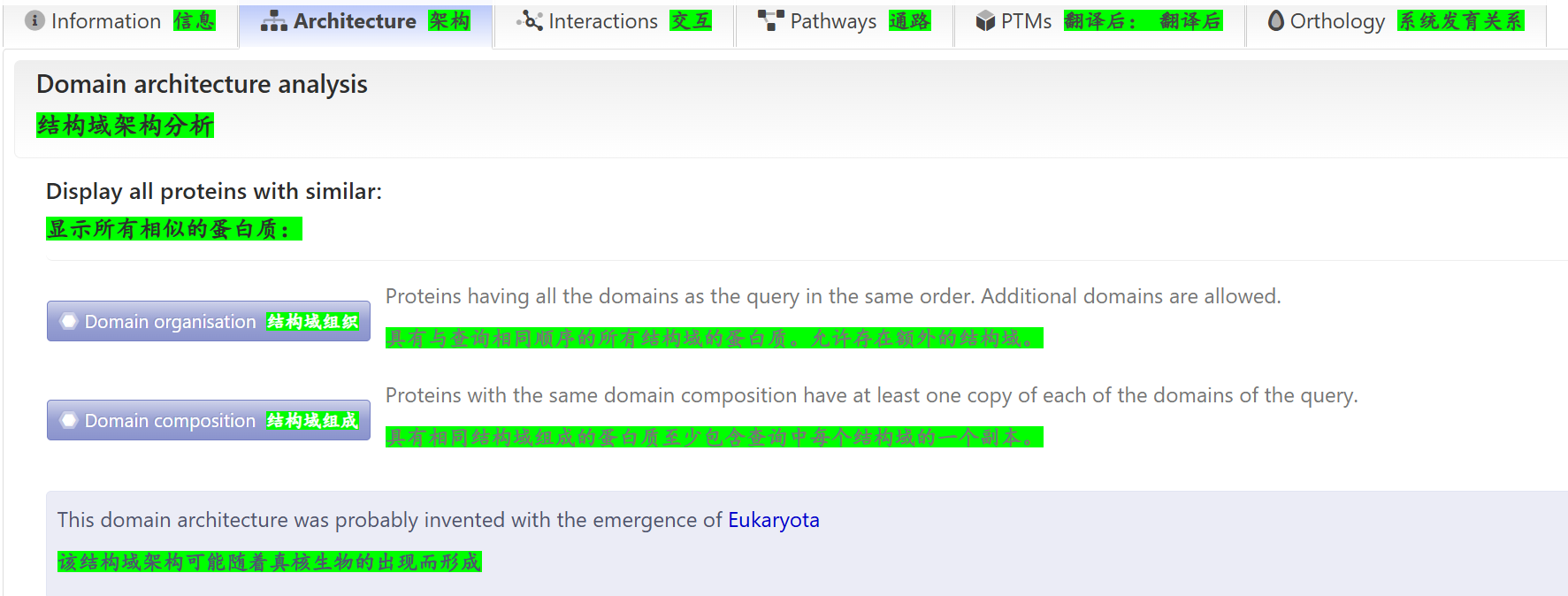
蛋白质互作分析:
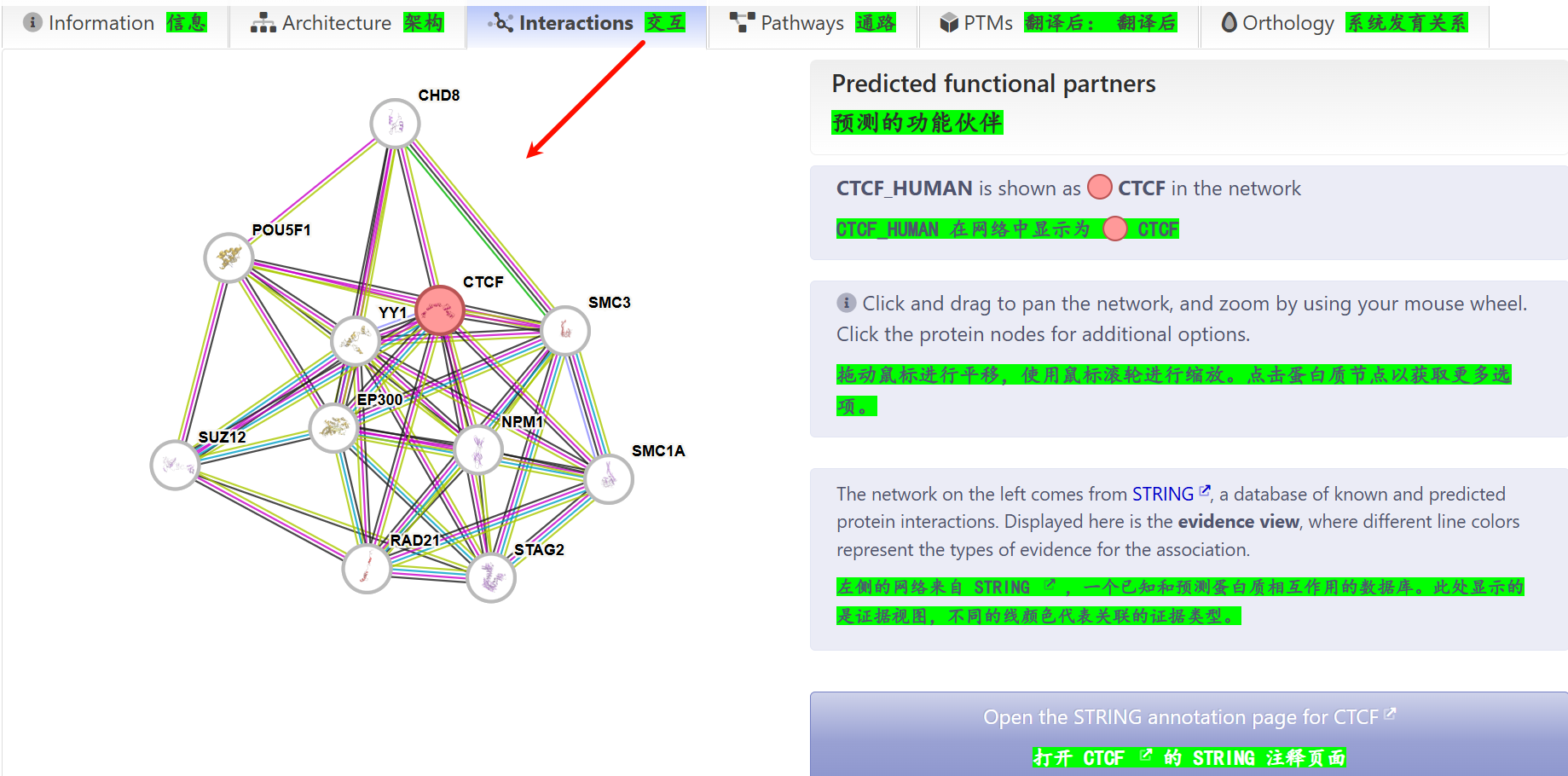
这个其实就没什么好说的了,也是数据库套娃,援引了STRING蛋白质互作(PPI)数据库,后者是大家都熟悉的。
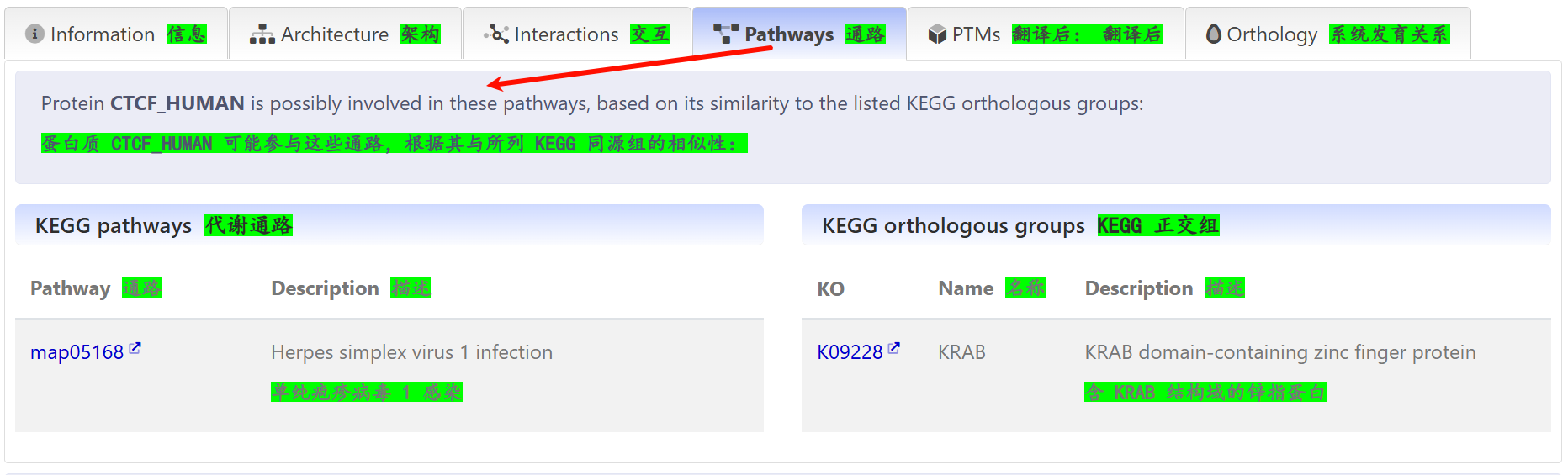
通路分析
这个就一般般了,而且看着也没有KEGG、GO那样规范,暂时没有深入。
PTM翻译后修饰:

这个其实有很多工具、数据库都有做,
Uniprot界面其实也有专门收集翻译后修饰位点+文献参考的条目:
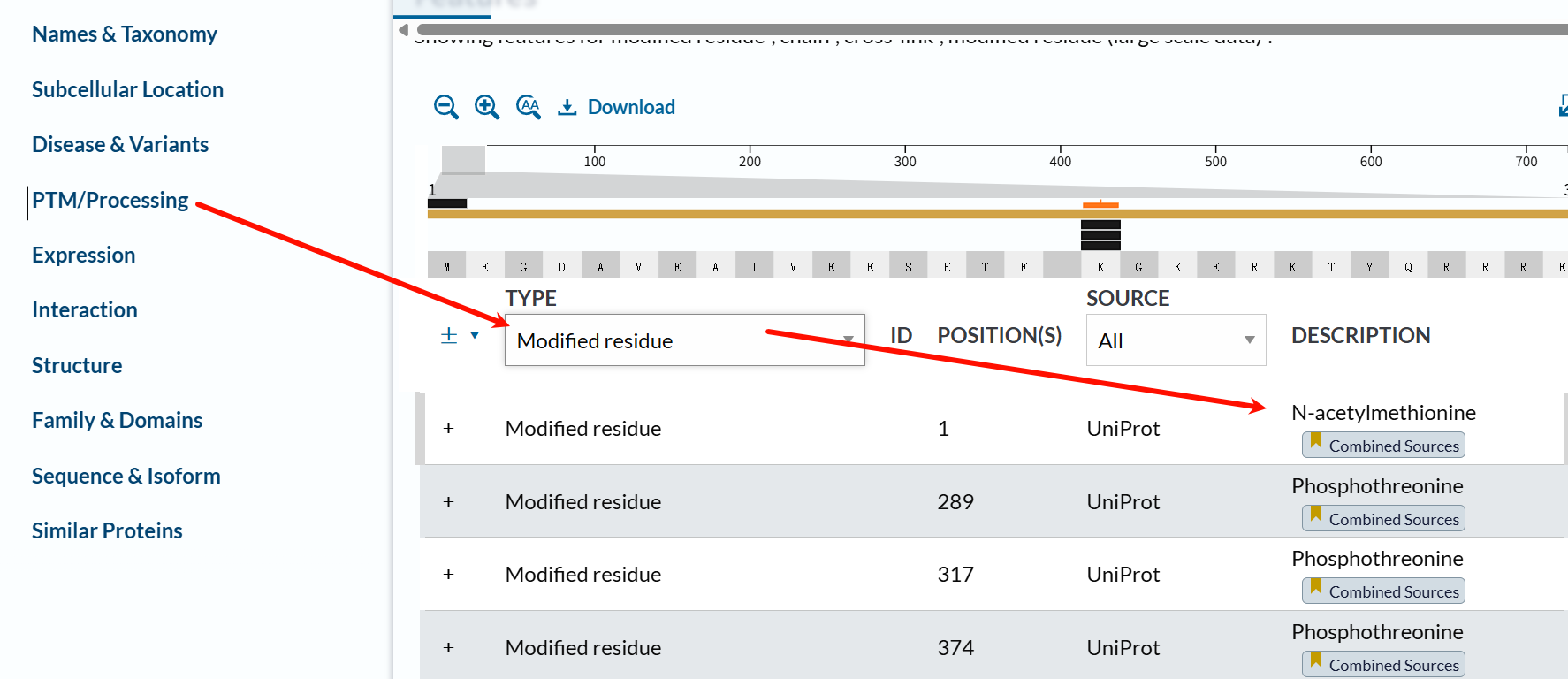
在描述里能够看到具体是什么修饰,以及该修饰的数据来源,也就是文献参考。
文献有好有坏,有些是生化实验(就是手工作坊那一套,一个一个精修发现的),有些修饰就是高通量蛋白质组学实验获取的数据,
而且预测修饰的深度学习工具,现在也有很多,要注意是实际修饰存在(什么细胞中),还是经过预测获得的数据。
关于如何获取Uniprot中蛋白质的PTM数据的脚本化方法,可以参考之前的博客:
系统发生分析(进化分析)


能够看到是按照分类单元一层一层往下,直到CTCF human。
12,SUPERFAMILY(https://supfam.org/SUPERFAMILY/)


主要功能:






SUPFAM提供的web server:
参考:https://supfam.org/SUPERFAMILY/web_services.html


可以使用分布式注释系统请求,通过URL格式向SUPFAM等DAS服务器发起请求,获取生物序列相关的注释数据,无需下载安装数据库转储即可实时更新信息。
13,总结:从InterPro出发
其实看了一圈之后,个人感觉能用于底层预测的数据库,而非只是简单引用,主要有Pfam/Prosite/SMART。
而这些数据库其实都是InterPro的成员数据库,
那么我们的目的,就是使用InterPro数据库来访问,或者是预测我们需要的蛋白质结构域domain注释数据。
InterPro的文档参考:
https://interpro-documentation.readthedocs.io/en/latest/index.html
现在,我们回过头来,再正式地认识一下InterPro数据库:

可以看到,InterPr其实是一个蛋白质数据库联盟,集成了一共13个蛋白质分析相关的数据库(我们前面探究的其实只是我个人认为涉及到结构域的部分数数据库);

下面的这个图片其实就很明显了:
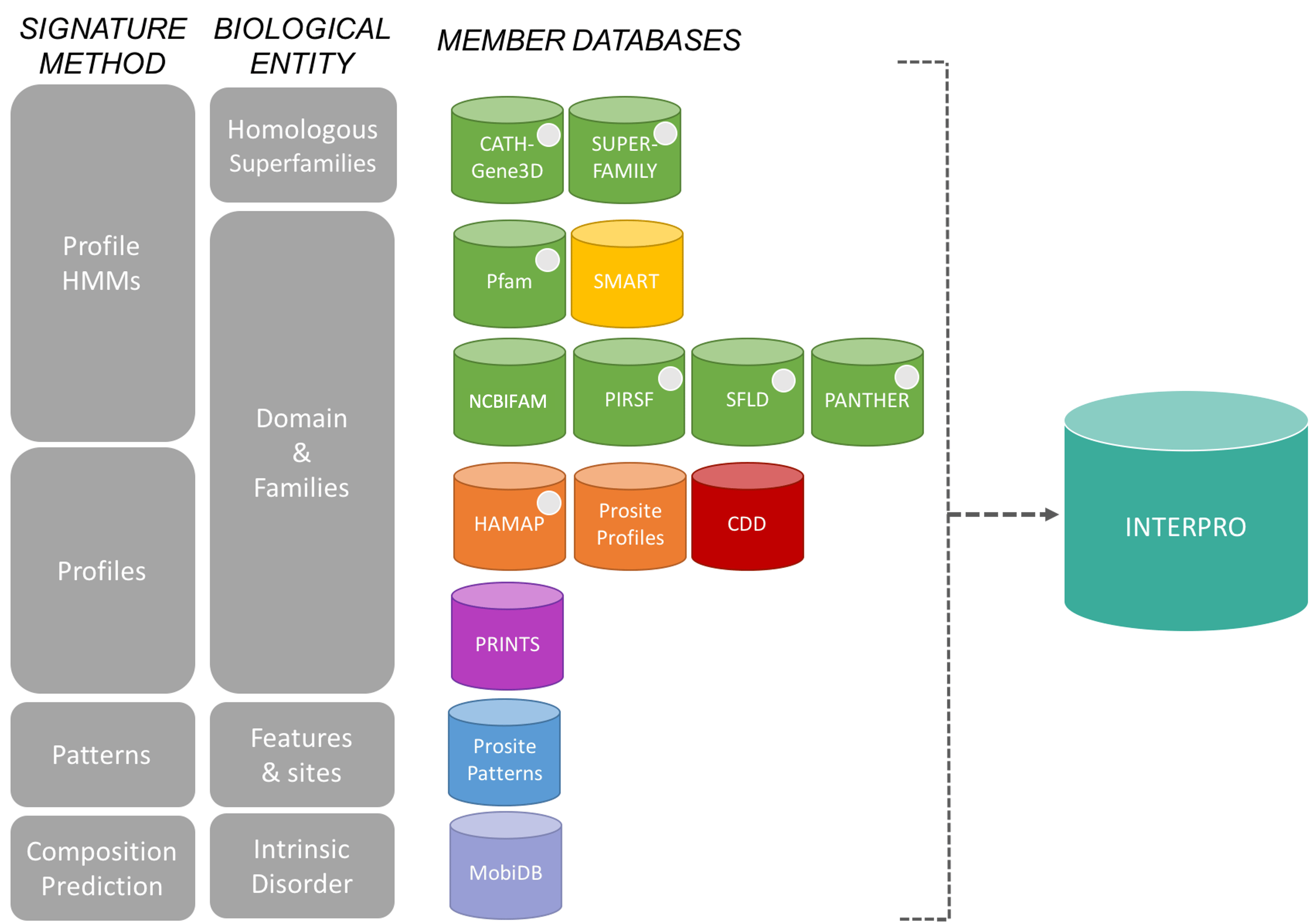

具体使用web server时展示的各种注释,参考:https://interpro-documentation.readthedocs.io/en/latest/protein_viewer.html
此处我们先展示一下效果:
还是以CTCF human为例,此处展示的是按照其uniprot id也就是P49711交叉参考的数据,
链接参考:https://www.ebi.ac.uk/interpro/protein/UniProt/P49711/
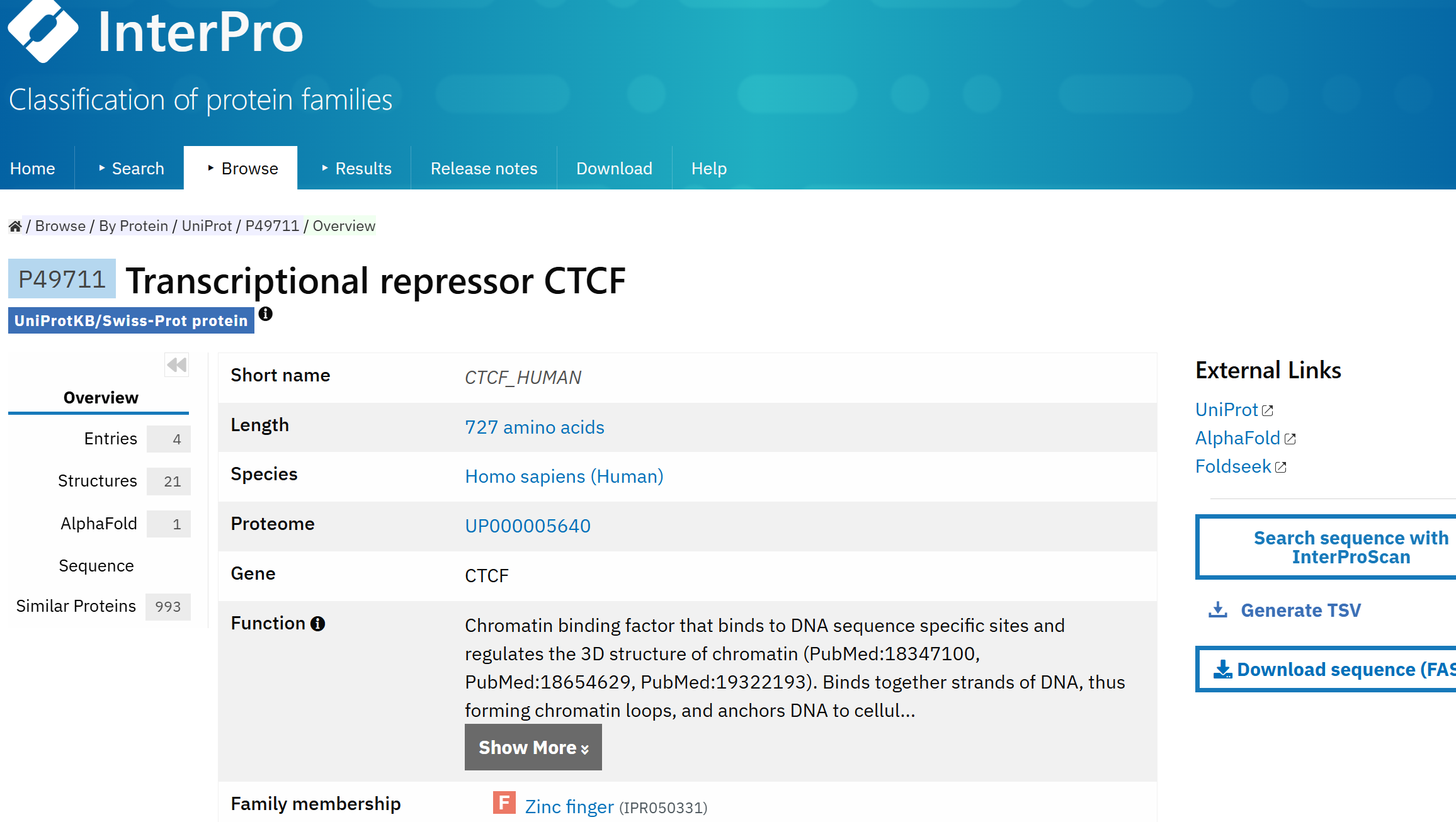
我们可以只看其中结构域部分数据:
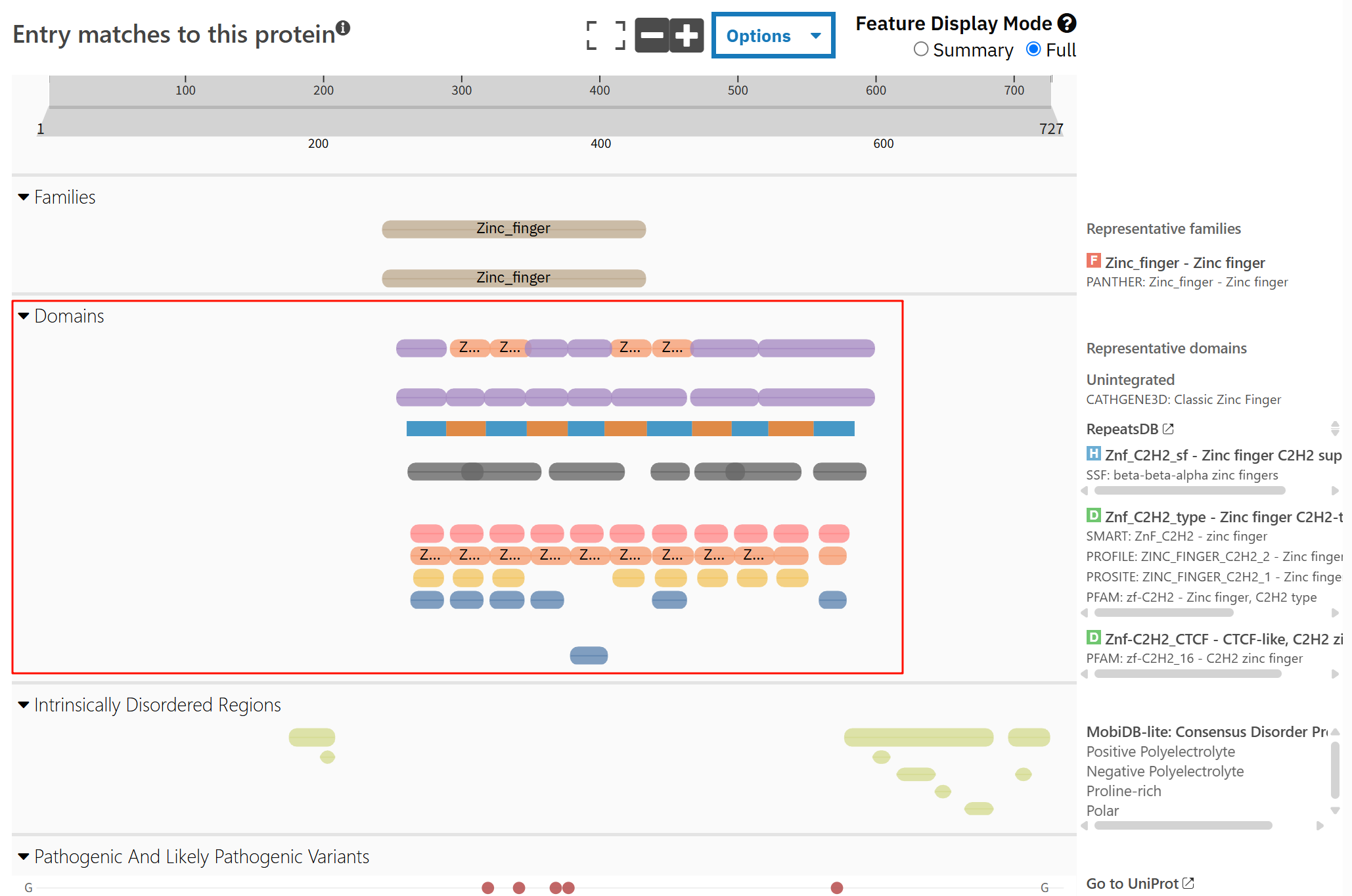
确实interpro就是整合其13个子数据库的数据,
而且我们肉眼不能够发现红框中domain部分,不同数据库注释出来结果有差异。
重点:如何使用&接口访问
我们的重点是如何使用、下载(并且最好是能够脚本化、整合进流程)InterPro数据,
参考链接:https://interpro-documentation.readthedocs.io/en/latest/download.html。
我们的目的更多的是,直接从InterPro API下载过滤后的数据,并集成到自己的程序中。
(1)InterProScan软件:
官方文档链接,参考:https://interpro-documentation.readthedocs.io/en/latest/interproscan.html

详细软件文档说明,参考:https://interproscan-docs.readthedocs.io/en/v5/

(2)InterPro API(应用程序编程接口)
官方仓库文档链接,见https://github.com/ProteinsWebTeam/interpro7-api/tree/master/docs
可以按照官方文档中的实例一步一步来实现。
(3)官方调用API的脚本生成器
参考链接:https://interpro-documentation.readthedocs.io/en/latest/faq.html

具体界面,参考:https://www.ebi.ac.uk/interpro/result/download/#/entry/InterPro/|accession
在前面设置完之后,会有一个code example代码示例,
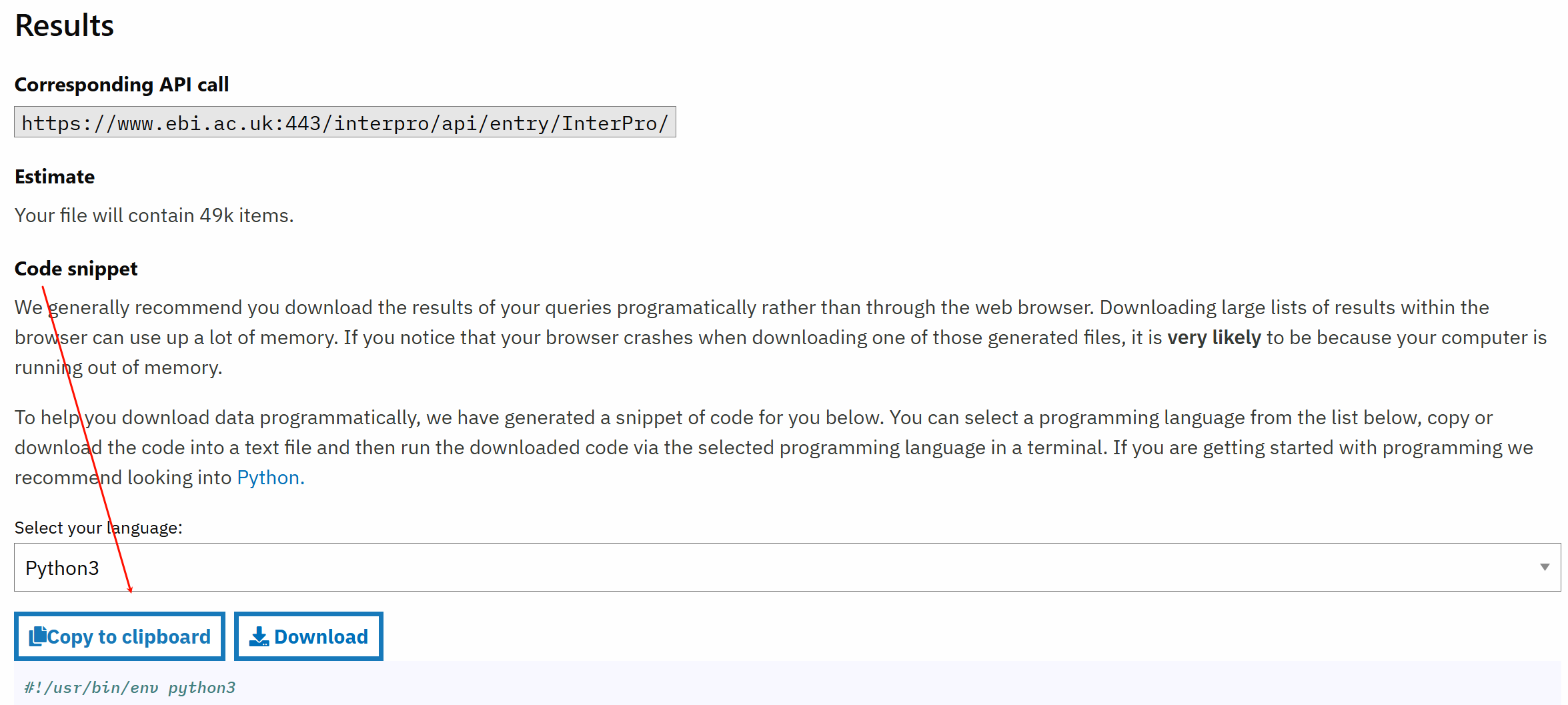
按照这个实例照葫芦画瓢,就可以批量处理自己的数据了,也就能够写出批量提取结构域的数据脚本了。
三,to-do
● (一)中,对于uniprot结构域数据的api query挖掘,参考:https://www.uniprot.org/help/sequence_annotation,以及如何通过xref补全其他数据库注释,主要是InterPro
● (二)中,对于InterPro数据库其实我没怎么讲API的调用,但这个数据库其实是比Uniprot更加丰富的,至少从结构域分析方面,另外InterProScan的示例我也还没有整理完毕