原文链接:使用R语言绘制SNP曼哈顿图
本期教程
文章来源

**文章:**Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber
**网址:**https://pmc.ncbi.nlm.nih.gov/articles/PMC8813957/
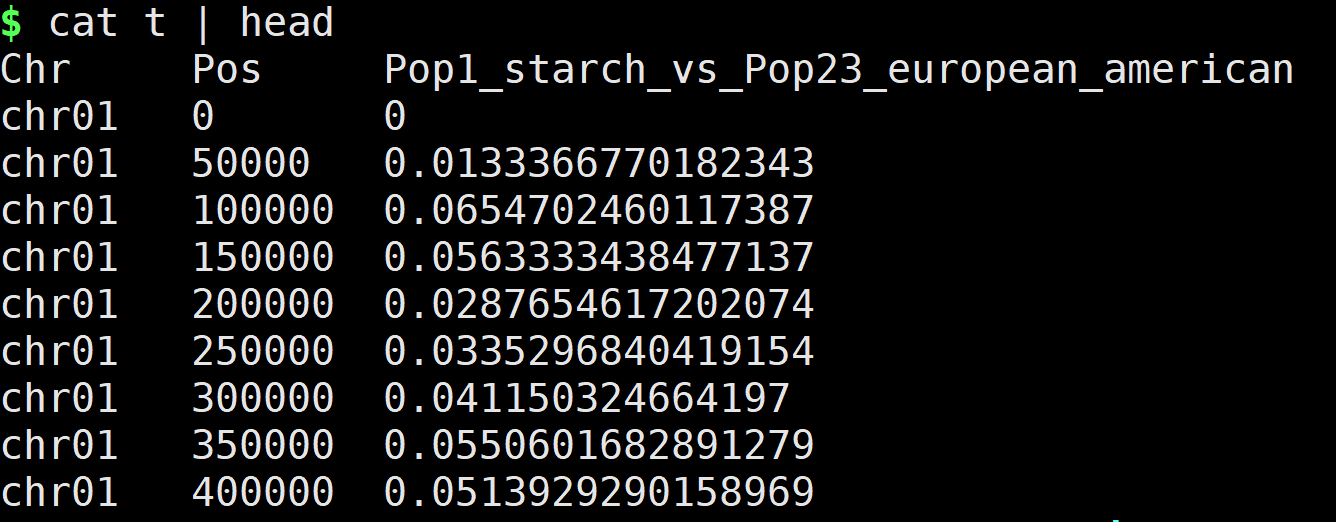
绘图数据
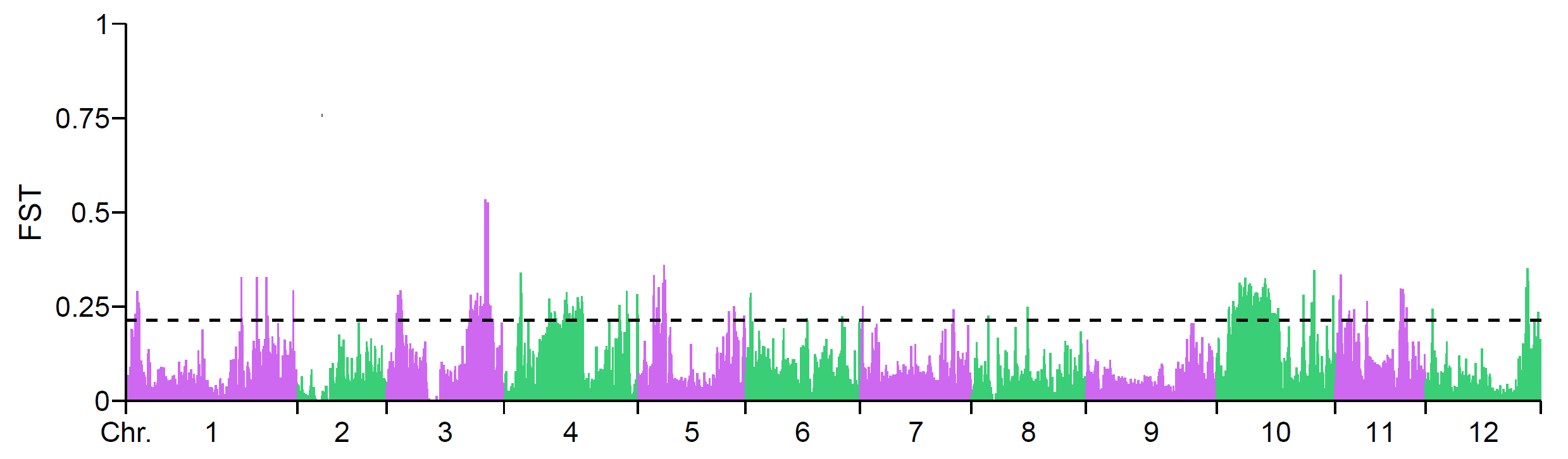
- 图展示了 基因组各染色体上成千上万个 SNP 位点的 FST 值分布
- FST是衡量两个群体遗传分化程度,数值越高表示差异越大
绘图代码
R
argv<-commandArgs(TRUE)
library('stringr')
chr_num=as.numeric(argv[3]) ### total number of chromosomes
chr_pre=argv[4] ### fasta header of reference chromosome
threshold=as.numeric(argv[5]) ### 0.2153, genomewide 5% threshold FST
pdf(argv[2], 10, 3)
par(mar=c(2,4,1,1),oma=c(0,0,0,0),mgp=c(2.5,0.5,0),cex.axis=1.1,las=1,cex.lab=1.2,lend=1)
a <- read.table(argv[1],header=T)
###Chr len for Pos infor
len <- c(0)
a[,1] <- as.factor(a[,1])
for (i in 1:chr_num) { #### chromosomes
snp <- subset(a,a$Chr==levels(a[,1])[i])
len <- c(len,snp[,2][length(snp[,2])])
}
plot(1,1,type='n',xaxs='i',yaxs='i',cex=0.1,col='steelblue1',axes=F,main='',xlab='',ylab='FST',xlim=c(0,sum(len)),ylim=c(0,1))
par(new=T)
len_sum <- 0
len_sum_d <- c(0)
for (i in 1:chr_num){ #### chromosomes
if (i %% 2 == 1){
color <- '#ce68f1'
} else {
color <- '#3ace76'
}
ii <- str_pad(i, 2, side='left', '0')
d <- subset(a,Chr == paste(chr_pre,ii,sep='')) #### chromosomes
len_sum = len_sum + len[i]
len_sum_d <- c(len_sum_d,len_sum)
pos <- (d$Pos)
#plot(len_sum+pos,d$Pop1_starch_vs_Pop23_european_american,type='h',xaxs='i',yaxs='i',col='gray81',axes=F,main='',xlab='',ylab='',xlim=c(0,sum(len)),ylim=c(0,7000)) # snp number distribution
#par(new=T)
for (n in 1:length(d[,2])){
plot(1,1,type='n',xaxs='i',yaxs='i',cex=0.1,col='blue',axes=F,main='',xlab='',ylab='',xlim=c(0,sum(len)),ylim=c(0,1)) # add layer out
points(len_sum+d[n,2],d[n,3],type='h',xaxs='i',yaxs='i',cex=0.2,col=color,pch=20) # plot point
# points(len_sum+d[n,2],d[n,7],type='p',xaxs='i',yaxs='i',cex=0.2,col='red',pch=20) # plot point
par(new=T)
}
par(new=T)
}
len_sum_d <-c(len_sum_d,sum(len))
len_sum_d1<-len_sum_d[2:length(len_sum_d)]
#axis(4,tcl=-0.42,lwd=2,lwd.ticks=2,at=seq(0,1,0.2),labels=seq(0,7000,1400))
#mtext('Number of variations', side = 4, line = 3.2,cex=1.3,las=0)
axis(2,tcl=-0.42,lwd=1,lwd.ticks=1,at=seq(0,1,0.25),labels=seq(0,1,0.25))
axis(1,at=c(len_sum_d1[1],len_sum_d1[2],len_sum_d1[3],len_sum_d1[4],len_sum_d1[5],len_sum_d1[6],len_sum_d1[7],len_sum_d1[8],len_sum_d1[9],len_sum_d1[10],len_sum_d1[11],len_sum_d1[12],len_sum_d1[13]),labels=F,lwd=1,lwd.ticks=1,tcl=-0.42) #### chromosomes
axis(1,tick=F,at=c(0,len_sum_d1[2]/2,len_sum_d1[2]+(len_sum_d1[3]-len_sum_d1[2])/2,len_sum_d1[3]+(len_sum_d1[4]-len_sum_d1[3])/2,len_sum_d1[4]+(len_sum_d1[5]-len_sum_d1[4])/2,len_sum_d1[5]+(len_sum_d1[6]-len_sum_d1[5])/2,len_sum_d1[6]+(len_sum_d1[7]-len_sum_d1[6])/2,len_sum_d1[7]+(len_sum_d1[8]-len_sum_d1[7])/2,len_sum_d1[8]+(len_sum_d1[9]-len_sum_d1[8])/2,len_sum_d1[9]+(len_sum_d1[10]-len_sum_d1[9])/2,len_sum_d1[10]+(len_sum_d1[11]-len_sum_d1[10])/2,len_sum_d1[11]+(len_sum_d1[12]-len_sum_d1[11])/2,len_sum_d1[12]+(len_sum_d1[13]-len_sum_d1[12])/2),labels=c('Chr.', '1','2','3','4','5','6','7','8','9','10','11','12')) #### omosomes
#for (i in 2:chr_num){ #### chromosomes
# segments(len_sum_d1[i],-1,len_sum_d1[i],1,lty=2,lwd=1.35,col=rgb(0,0,0,0.5))
#}
abline(h=threshold,lty=2,lwd=1.6,col='black')
#legend('topright',legend='95% confidence interval',col='red',bty='n',lty=1,lwd=2,cex=1)运行:
R
Rscript ./plot_fst.R t 166_potato_starch_vs_others_Fst.pdf 12 chr 0.2135参数解释
| 参数位置 | 示例值 | 可能含义(常见脚本格式) |
|---|---|---|
| 1 | t |
输入数据文件前缀或模式(例如 t.Fst.stat) |
| 2 | 166_potato_starch_vs_others_Fst.pdf |
输出 PDF 文件名(最终图像文件) |
| 3 | 12 |
染色体数量(如马铃薯 12 条染色体) |
| 4 | chr |
染色体列名称前缀(例如 chr1, chr2...) |
| 5 | 0.2135 |
FST 阈值(画虚线用的 cutoff 值) |
若我们的教程对你有所帮助,请点赞+收藏+转发,大家的支持是我们更新的动力!!
2024已离你我而去,2025加油!!
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。)
2. 精美图形绘制教程
3. 转录组分析教程
4. 转录组下游分析
BioinfoR生信筆記 ,注于分享生物信息学相关知识和R语言绘图教程。