摘要:掌握Linux命令行操作是生物信息分析的基石。本文为您梳理必须掌握的核心Linux命令,并详细解释它们在生物信息学中的典型应用场景,帮助您快速上手生物信息分析。
一、核心理念:Linux是工具,数据处理是目的
不要被命令吓倒。您的目标不是成为系统管理员,而是学会如何使用这些工具来处理测序数据、基因组、注释文件等。学习模式应为:知道有什么工具 -> 在何时使用 -> 如何组合使用。
见末尾,推荐一个网站,里面有详细的命令说明,很方便随时查询。
二、第一阶段:生存技能 - 基础导航与文件操作
这是您登录服务器后,首先要会用的命令。
1. pwd (Print Working Directory)
-
作用:显示当前所在目录的绝对路径。
-
场景:在复杂的目录结构中迷路时,快速定位自己在哪里。
2. ls (List)
-
作用:列出目录中的文件和子目录。
-
关键选项:
-
-l:以长格式显示(权限、所有者、大小、时间) -
-h:与-l结合,将文件大小以易读格式(K,M,G)显示 -
-a:显示所有文件,包括隐藏文件(以.开头的文件)
-
-
示例:
ls -lh /home/user/data/ # 查看数据目录的详细内容
3. cd (Change Directory)
-
作用:切换目录。
-
关键用法:
-
cd ~或cd:回到用户家目录 -
cd ..:返回上一级目录 -
cd -:在两个目录间快速切换
-
4. mkdir / rm (Make/Remove Directory)
-
作用:创建/删除空目录。
-
关键选项 :
mkdir -p可以创建多级嵌套目录 -
示例:
mkdir -p project/{raw_data,scripts,results}
5. cp / mv / rm (Copy/Move/Remove)
-
作用:复制、移动(重命名)、删除文件和目录。
-
⚠️ 极度重要警告:Linux命令行下删除操作不可逆!没有"回收站"!
-
安全习惯:
-
对
rm总是先使用ls确认要删除的文件 -
使用
rm -i在删除前询问确认 -
对于目录,使用
rm -r(递归删除)。慎用rm -rf(强制递归删除)!
-
6. cat / less / head / tail (查看文件内容)
-
cat:快速查看小文件内容,或用于文件合并、重定向
-
less 【最常用】:分页查看大文件。支持上下翻页、搜索(按
/然后输入关键词)。按q退出 -
head / tail:查看文件开头或结尾的N行(默认10行)
三、第二阶段:核心战斗力 - 文本处理与搜索
生物数据大多是文本文件(FASTA、FASTQ、GTF、SAM、CSV等),以下命令是您的"手术刀"。
1. grep (Global Regular Expression Print)
-
作用:在文件中搜索指定的字符串或模式(正则表达式)
-
生物信息学应用:从注释文件中提取特定基因的行、在日志中查找错误、统计FASTA文件中序列ID等
-
关键选项:
-
-i:忽略大小写 -
-v:反向搜索,输出不匹配的行 -
-c:只统计匹配的行数 -
-E:启用扩展正则表达式 -
-w:匹配整个单词
-
-
示例:
grep -c "^>" genome.fa # 统计FASTA文件中的序列条数(以>开头的行) grep -c 'R' plink.fam # 统计plink.fam文件中包含'R'的行数
2. awk
-
作用:强大的文本分析工具,擅长处理表格数据(以空格、制表符分隔的列)
-
基本语法 :
awk '模式 {动作}' 文件 -
生物信息学应用:提取某一列、根据条件过滤行、简单计算
-
内置变量:
-
$1, $2, ... $NF:表示第1,2,...最后一列 -
NR:表示行号
-
-
示例:
# 打印第1列和第3列,用制表符分隔 awk '{print $1, "\t", $3}' input.txt # 输出第5列值大于0.05的所有行 awk '$5 > 0.05 {print $0}' differential_genes.txt # 计算第2列的平均值 awk '{sum+=$2} END {print sum/NR}' data.txt
3. sed (Stream Editor)
-
作用:流编辑器,用于对文本进行查找、替换、删除、插入等操作
-
生物信息学应用:批量修改文件名(配合循环)、清理数据格式、替换字符
-
经典示例:
sed 's/old_string/new_string/g' input.txt > output.txt-
将文件中所有
old_string替换为new_string -
s表示替换,g表示全局(如批量替换染色体编号)
-
4. cut / paste / sort / uniq / wc
-
cut:按列切割文件
cut -f 1,3 -d "," file.csv # 用逗号分隔符,取第1,3列 -
sort:排序
-
-n:按数字排序 -
-k:指定排序列
-
-
uniq:去除相邻的重复行。常与 sort 连用:
sort file.txt | uniq -c # 统计每个唯一项的出现次数 -
wc:统计行数、词数、字节数
wc -l huge_file.fastq # 快速知道你有多少条测序序列(行数/4)
四、第三阶段:效率与流程 - 软件、权限与进程管理
1. 软件安装与管理 (which, 包管理器)
-
which:查看某个命令或软件的安装路径
which bwa -
生物信息学推荐:使用 conda (特别是 mamba) 来管理生物信息学软件和环境,它可以解决令人头疼的依赖问题(这些暂时接触不到,也可以先不用,后面有机会再写)
-
下载
wget -c https://repo.anaconda.com/archive/Anaconda3-2021.05-Linux-x86_64.sh
bash Anaconda3-2021.05-Linux-x86_64.sh2. 文件权限 (chmod, chown)
-
chmod:修改文件权限。需要知道如何给脚本添加可执行权限:
chmod +x my_script.sh
3. 进程管理 (ps, top, kill, nohup, &)
-
&:在命令末尾添加,让任务在后台运行
-
nohup:让任务在您退出登录后继续运行
nohup command > output.log 2>&1 & -
ps / top:查看进程状态和资源占用
-
kill:终止进程
kill -9 PID # 强制终止
4. 输入/输出重定向与管道 (>, >>, |)
-
>:将标准输出重定向到文件(覆盖)
-
>>:将标准输出追加到文件
-
| (管道):【灵魂所在】将一个命令的输出作为下一个命令的输入
-
生物信息学管道示例:
zcat reads.fastq.gz | head -n 400000 # 解压、取前40万条序列、进行质控
五、学习路径建议
-
边用边学:不要死记硬背所有参数。从一个实际任务开始,例如:"我想看看我的测序数据(FASTQ文件)前几条序列长什么样"
-
组合使用 :练习将
grep,awk,sort,uniq用管道连接起来,解决复杂问题 -
善用帮助:
-
man command(如man grep)查看官方手册 -
command --help或-h查看快速帮助
-
-
编写Shell脚本 :当你需要重复执行一系列命令时,就是学习编写 bash 脚本 (
*.sh) 的时候了。这是实现分析流程自动化的第一步
六、一个经典的综合示例
统计一个文件多少条染色体,以及每条染色体有多少条标记的分布:
# 假设文件是 dp4miss0.2.bim
# 1. 提取染色体
# 2. 计算每行染色体出现次数
# 3. 排序
# 4. 统计每个染色体出现的次数
cut -f 1 dp4miss0.2.bim |sort|uniq -c > chr.list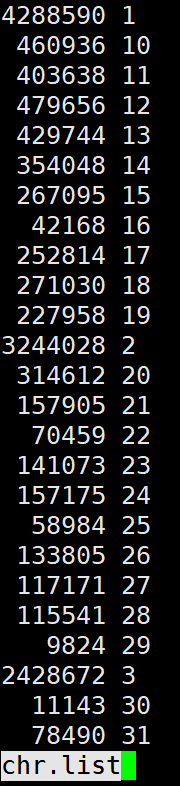
七、实战数据集推荐与练习
对于生物信息学初学者,推荐从以下类型的简单数据集开始练习:
1. FASTA格式数据(适合练习grep、awk)
-
来源:从NCBI下载
-
如手动下载
-
https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_029584285.1/
-
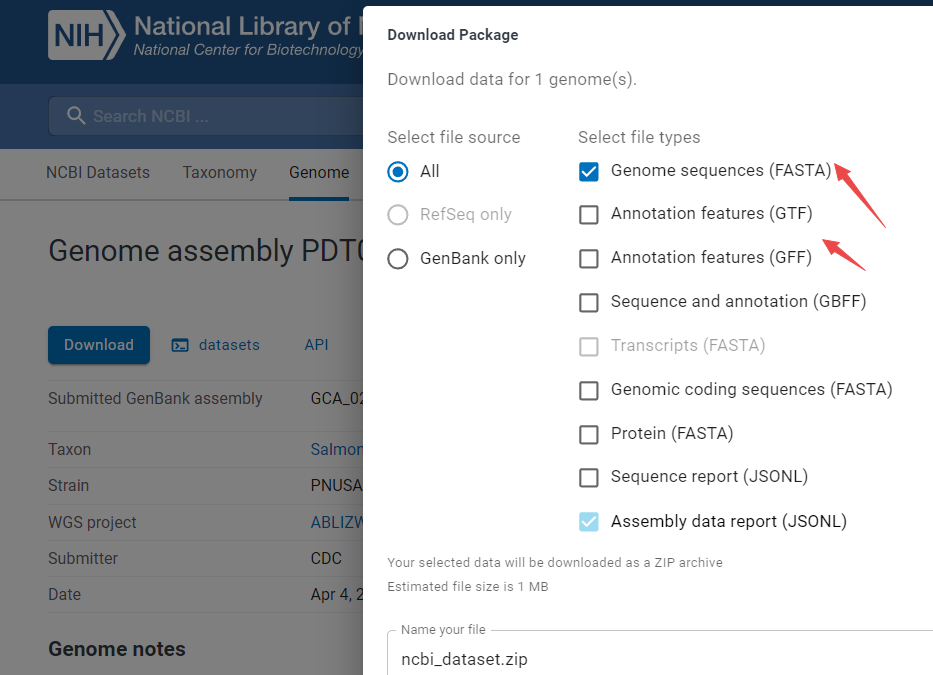
-
或者Ensembl Plants:
-
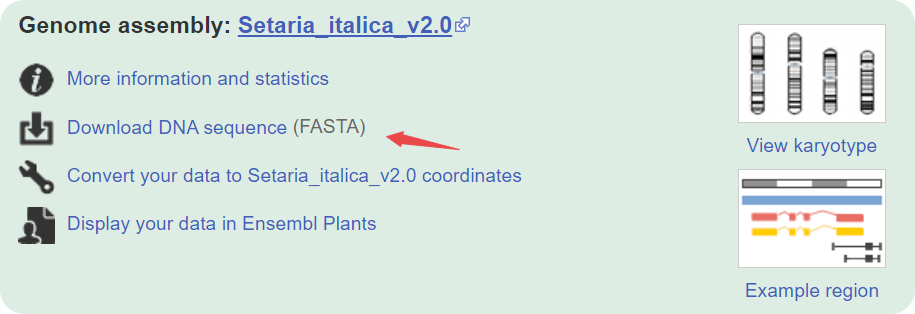
-
或者脚本下载,重要命令wget
-
练习任务:
grep -c "^>" genome.fa # 提取所有序列ID grep "^>" genome.fa | sed 's/>//g' # 计算总碱基数 grep -v "^>" genome.fa | tr -d '\n' | wc -c #结果 grep -c "^>" genome.fa 9 grep "^>" genome.fa | sed 's/>//g' 1 2 3 4 5 6 7 8 9 grep -v "^>" genome.fa | tr -d '\n' | wc -c 422473852
2. FASTQ格式数据(适合练习awk、管道)
-
来源 :GEO获取访问:https://www.ncbi.nlm.nih.gov/geo/
-
下载流程: 用到我们之前提到的wget
-
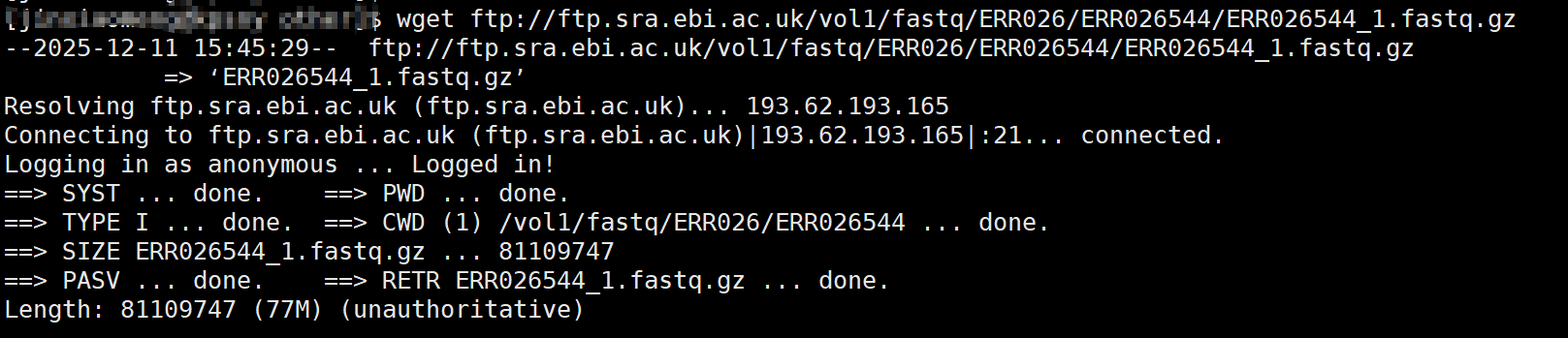
-
练习任务:
-
样本:ERR026544(金黄色葡萄球菌测序数据)
-
平台:Illumina
# 1. 下载文件
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR026/ERR026544/ERR026544_1.fastq.gz
# 2. 查看基本信息
ls -lh ERR026544_1.fastq.gz
# 输出:-rw-r--r-- 1 user group 2.5M ERR026544_1.fastq.gz
# 3. 解压(可选,很多工具可以直接处理gzip压缩文件)
gunzip ERR026544_1.fastq.gz
# 4. 查看文件内容
head -n 4 ERR026544_1.fastq
# 或不解压直接查看
zcat ERR026544_1.fastq.gz | head -n 4
# 5. 统计序列数
echo "序列数: $(( $(wc -l < ERR026544_1.fastq) / 4 ))"
# 或不解压统计
zcat ERR026544_1.fastq.gz | awk 'END{print NR/4}'3. 注释文件(GTF/GFF格式,适合练习cut、sort、uniq)
-
来源:Ensembl或UCSC的基因注释文件,同上面
-
练习任务:
# 统计不同特征类型 cut -f 3 annotation.gtf | sort | uniq -c # 提取特定染色体的注释 grep "^chr1\t" annotation.gtf > chr1_annotation.gtf # 统计每个基因的外显子数 awk '$3=="exon" {print $10}' annotation.gtf | sort | uniq -c
八、常见问题与解决方案
Q1:如何查找命令的使用方法?
# 方法1:使用man手册
man grep
# 方法2:查看帮助信息
grep --help
# 或
grep -hQ2:如何查看文件的前几行和后几行?
# 查看前5行
head -n 5 file.txt
# 查看后5行
tail -n 5 file.txt
# 同时查看首尾
(head -n 10; tail -n 10) < file.txtQ3:如何查找特定进程并终止?
# 终止进程(假设PID为12345)
kill 12345
# 强制终止
kill -9 12345九、总结与进阶建议
最后,也是最重要的:勤于练习,勇于尝试(在非重要数据上)。遇到错误信息时,仔细阅读,并善用搜索引擎(错误信息就是关键词),这是解决问题的关键能力。
记住,在生物信息学中,Linux命令行是你的超级工具箱。每个命令都是一个工具,而真正的艺术在于知道如何组合这些工具来解决具体的生物学问题。
祝您在生物信息学的学习之旅中顺利!
提示:本文提供的所有命令示例都经过简化,实际使用时请根据具体需求调整参数。建议在虚拟机或测试服务器上练习这些命令,避免在生产环境中误操作。
在下一期文章中,我们将深入探讨:
《生物信息学常用数据库完全指南》将学到:NCBI(美国):SRA、GEO
相关资源: