AI 制药编码之战:预测癌症反应,选对方法是关键

目录
- 预测癌症对药物或基因编辑的反应,关键在于为基因、化合物和细胞系选择最佳的「翻译」方法;这项研究表明,STRING、MiniMol 和原始基因表达数据分别在各自领域胜出。
- MolGuidance 通过混合引导策略,揭示了原子类型和化学键等离散特征在调控分子性质上比三 - 维空间结构更关键,实现了前所未有的生成精度。
- 通过检索相似反应来「提醒」AI,RARB 框架显著提升了逆合成路线预测的准确性和泛化能力。
- 这项研究构建了高性能聚合物基础模型,但意外发现模型可能依赖数据插值而非理解化学,警示我们需谨慎评估 AI 的「理解力」。
- EXAONE Path 2.5 通过巧妙融合病理图像与多组学数据,为精准肿瘤学构建了更具生物学意义的患者模型。
- HeMeNet 模型通过多任务学习和全原子图表示,让 AI 在蛋白亲和力等多个预测任务上表现出色,为解决药物研发中数据稀疏问题提供了新思路。
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-QGFTayN5-1766808738057)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
1. AI 制药编码之战:预测癌症反应,选对方法是关键
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-VatyAeKo-1766808738057)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
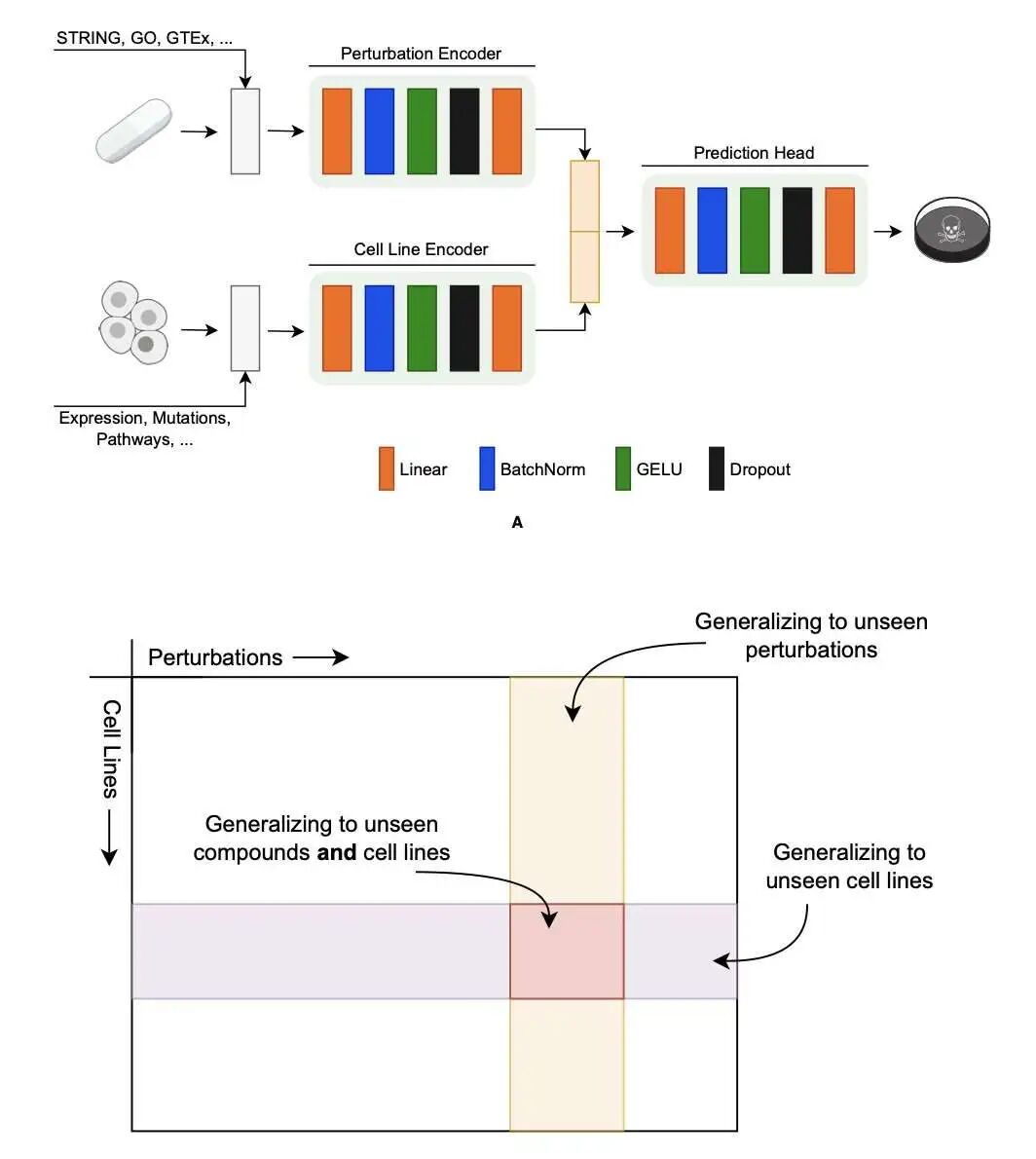
在精准医疗领域,我们一直想回答一个核心问题:哪种药对哪种癌细胞最有效?为了找到答案,我们建立了庞大的数据库,比如 DepMap 和 PRISM,记录了成千上万种细胞系在面对各种药物(化学扰动)或基因敲除(遗传扰动)时的反应。有了数据,下一步就是建立预测模型。而所有模型的第一步,也是最关键的一步,就是如何将基因、化合物和细胞系这些生物化学实体「翻译」成机器能理解的语言。这个翻译过程,我们称之为「编码」(Encoding)。
选错了编码方式,就像给了机器一本错的词典,后续的一切分析都可能是徒劳。最近这篇论文就做了一件极其有价值的事:它系统性地比较了市面上各种主流的编码方法,像是一场编码界的「大乱斗」,帮我们找出谁才是真正的赢家。
基因编码:网络关系胜过一切
当我们要预测敲除某个基因会产生什么后果时,如何向模型描述这个基因?最直接的方法是看它的功能分类(比如 GO-term),或者用现在很火的蛋白质语言模型(Protein Language Models)给它生成一个「数字指纹」。
但研究者发现,效果最好的编码方式是 STRING。STRING 本质上是一张巨大的蛋白质相互作用网络图,它告诉我们一个蛋白质在细胞里都和谁「来往」。用 STRING 来编码一个基因,实际上是在描述它在整个社交网络中的位置和关系。这个结果说明,一个基因的功能,很大程度上是由它的「朋友圈」决定的。相比之下,其他编码方式更像是只描述了这个基因的个人简历,忽略了它的社会关系,因此信息量就差了一大截。
更有趣的是,研究者尝试将多种基因编码器组合起来,以为能「三个臭皮匠赛过诸葛亮」,结果发现性能提升微乎其微。这说明 STRING 提供的网络信息已经足够丰富,其他编码方式能提供的新信息非常有限。
化合物编码:领域知识是王道
对于化合物,我们通常用化学指纹(fingerprints)或者分子图(molecular graphs)来描述其结构。但这次的赢家是 MiniMol。
MiniMol 的秘诀在于「预训练」(pre-training)。它不是在一个通用的化学分子数据库上学习,而是在与药物研发直接相关的生物测定数据(bio-assay data)上进行训练。这就像教一个学生,不是让他去读一本通用百科全书,而是直接给他看大量真实的病例报告。这样训练出来的模型,自然更懂得如何从化学结构中解读出它可能产生的生物效应。这个发现告诉我们,对于化学分子编码,脱离生物学背景的纯化学描述,其预测能力是有限的。上下文(Context)至关重要。
细胞系编码:大道至简
最令人意外的发现可能来自细胞系的编码。我们通常认为,细胞系的状态非常复杂,应该用复杂的模型来描述。比如,现在有专门的转录组学基础模型(transcriptomics foundation model),试图从海量的基因表达数据中学习出一种更深层次、更抽象的细胞状态表示。
然而,研究结果却「返璞归真」:直接使用原始的基因表达谱数据,效果居然最好。它打败了那些经过复杂处理和抽象的编码方式,也优于基于基因型(genotype)的编码。
这说明什么?这说明对于预测扰动反应这件事,原始基因表达数据中那些最直接、最微妙的信号可能非常关键。复杂的模型在试图「降噪」和「抓重点」的过程中,可能不小心把这些关键信号也一并丢掉了。这就像看一张高清照片,我们能捕捉到很多细节;如果把它转换成一幅卡通简笔画,虽然轮廓还在,但大量信息已经丢失。在某些任务上,这些丢失的细节恰恰是决定成败的关键。
这项研究为所有从事相关模型开发的人员提供了一份非常实用的操作指南。它清楚地表明,不存在一种通用的「万能编码器」。针对不同的生物问题(基因、化合物、细胞系),我们需要选择最合适的工具。
📜Title: Benchmarking Chemical, Genetic, and Cell Line Encodings for Cancer Perturbation Response Prediction
🌐Paper: https://www.biorxiv.org/content/10.64898/2025.12.15.694331v1
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-qTzZTy7o-1766808738058)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
2. MolGuidance: AI 分子生成新突破,精准调控分子性质
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-afQf2lOk-1766808738058)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
做计算化学和药物设计,我们都有一个共同的目标:按需设计分子。就像给计算机一个指令,「我想要一个 HOMO-LUMO 能隙在特定范围的分子」,然后它就能直接生成结构。听起来很简单,但实现起来很难。现有的生成模型在「按需定制」这个环节,总是有点力不从心。
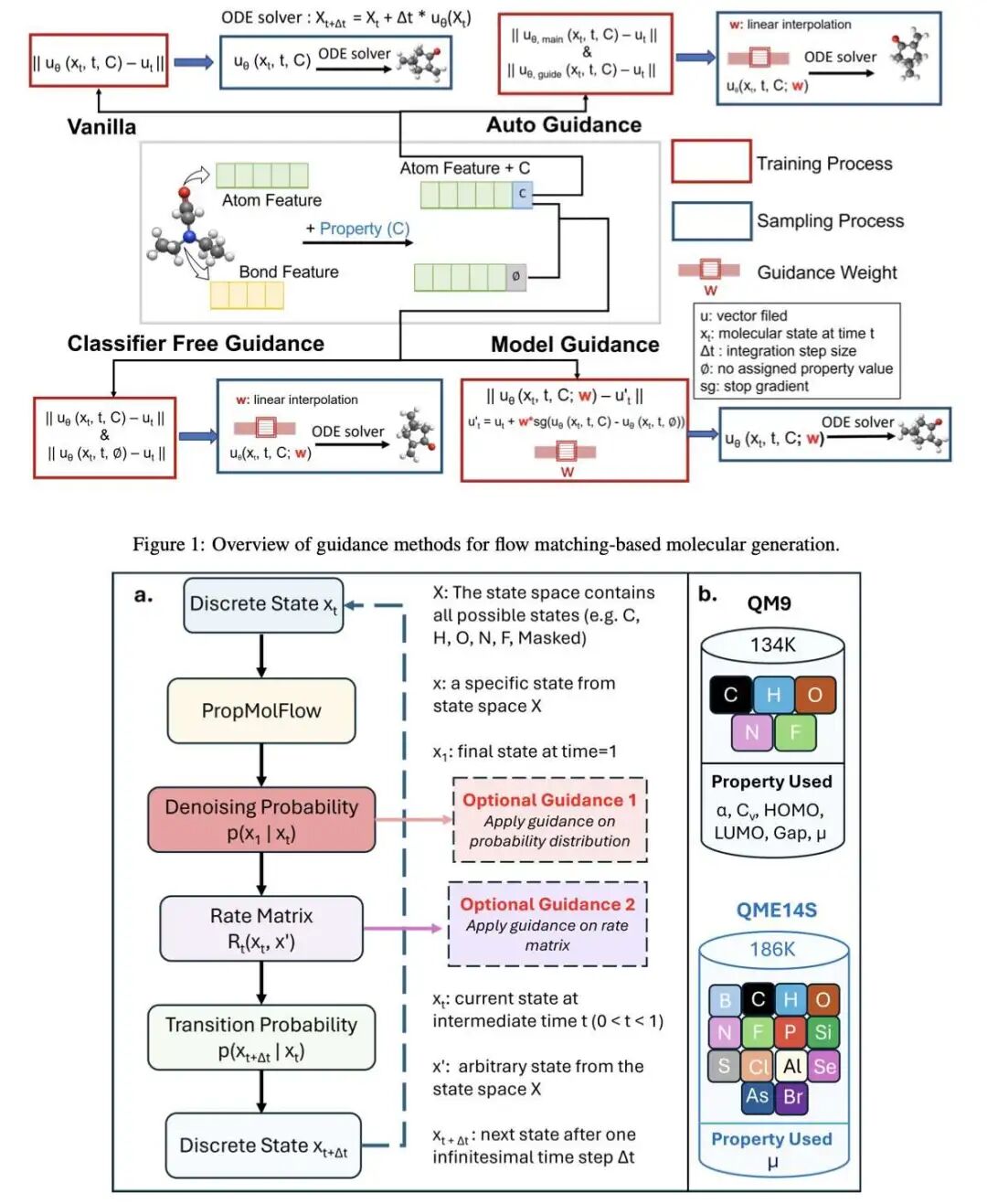
最近,一篇名为 MolGuidance 的论文,给我们带来了新的思路。作者们基于流匹配 (Flow Matching) 框架,开发了一套新的分子生成方法。流匹配本身就很强大,你可以把它想象成一个平滑的转换过程,把一团随机的噪声,一步步「雕刻」成一个结构清晰、化学合理的分子。它在保证生成多样性和新颖性方面,比扩散模型更有优势。
但这篇文章真正的亮点,在于他们对「引导」 (Guidance) 策略的深入研究。当我们需要生成具有特定性质的分子时,就需要一个「引导」来告诉模型该往哪个方向走。
研究者们发现了一个非常反直觉的现象。我们通常认为,分子的三维空间构象(也就是原子的坐标)对性质影响巨大,因此在模型训练时会投入大量计算资源去学习这种 SE(3) 等变性。但 MolGuidance 的实验表明,对于分子性质的调控,真正起决定性作用的,反而是那些离散的特征,比如原子是什么类型(碳、氧还是氮?)、化学键是单键还是双键。
这就像烤蛋糕。我们过去可能花了很多精力去设计一个形状完美的烤盘(三维几何),以为这样就能烤出美味的蛋糕。结果发现,真正决定蛋糕味道的,其实是你放了糖还是盐(原子类型和化学键)。
搞清楚了这一点,接下来的事情就顺理成章了。作者们设计了一套混合引导策略。他们把几种主流的引导方法,比如无分类器引导 (classifier-free guidance)、自引导 (autoguidance) 和模型引导 (model guidance),都整合了进来。
这个过程可以这么理解:
MolGuidance 的聪明之处在于,它没有偏信任何一种方法,而是用贝叶斯优化来动态地寻找一个最佳的组合权重。它知道什么时候该听导航的微调,什么时候该听向导的大方向指引。
为了验证这个方法到底行不行,作者们做了大量的实验。他们不只是生成了分子,还用密度泛函理论 (DFT) 这种计算化学的「金标准」方法,去实打实地计算生成分子的性质。结果显示,在 QM9 和 QMe14S 这些标准数据集上,MolGuidance 生成的分子在性质匹配度上,确实做到了当前最好。同时,生成的分子在化学有效性、稳定性和多样性方面也保持了高水准。
这项工作为我们精确设计具有特定功能的分子,打开了一扇新的大门。无论是在药物发现中寻找更高活性的先导化合物,还是在材料科学中设计具有特定光电性质的新材料,这种精准的「分子定制」能力,都是我们梦寐以求的。
📜Title: MolGuidance: Advanced Guidance Strategies for Conditional Molecular Generation with Flow Matching
🌐Paper: https://arxiv.org/abs/2512.12198v1
💻Code: https://github.com/Liu-Group-UF/MolGuidance
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-6Dd92YWu-1766808738058)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
3. 给 AI 配个检索库,逆向合成准确率提升 15%
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-8VMjwZE0-1766808738058)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
做逆合成分析,对人和 AI 来说都是个硬骨头。逆合成就像给你一把椅子,然后问你:「这东西最初是用哪几块木头、怎么拼起来的?」一个经验丰富的化学家会马上去翻文献,看看有没有人做过类似的椅子。这篇论文的作者们想,我们能不能也教 AI 学会这招?
绝大多数化学反应都不是把分子敲碎了从零开始,而是保留大部分核心骨架(scaffold),只在局部进行修饰。这意味着,如果我想合成一个新分子 A,那么去看看和它长得很像的分子 B 是怎么合成的,大概率能提供极好的线索。这套操作,就是化学家在实验室里用 SciFinder 或 Reaxys 查文献的日常。作者们做的,就是给 AI 模型内置了一个专属的「文献检索系统」。
这个新框架叫 RARB (Retrieval-Augmented RetroBridge),它的工作流程分为三步:
第一步是检索。你给模型一个目标产物,它的检索模块会立刻启动。这个模块使用摩根指纹 (Morgan fingerprints)------一种经典的分子结构编码方式------在庞大的反应数据库里,迅速找出几个和目标产物结构最相似的「亲戚」。这就像你想烤一个特别的蛋糕,就先去找几个外观最接近的食谱作参考。
第二步是信息整合。这是最巧妙的地方。系统不只是把找到的「参考食谱」一股脑丢给 AI,而是先用一个「信息提取器」把最有用的部分挑出来。这个提取器基于多头自注意力机制 (multi-head self-attention),也就是 GPT 这类大语言模型背后的核心技术。它能精准地识别出目标分子和参考分子之间的共同亚结构,以及反应可能发生的位点。然后,它把这些关键信息打包成一个简明的「提示 (prompt)」,交给下一步的生成模型。
第三步是生成。RARB 使用 RetroBridge 这个扩散模型 (diffusion model) 来具体生成反应物的结构。但和以前不同,它不再是蒙着眼睛瞎猜。它手里攥着上一步给的「提示」,就像拿到了一份详尽的备忘录,上面写着:「注意,分子的这部分骨架基本不会变,反应大概率发生在那边那个官能团上。」
结果怎么样?在标准的 USPTO-50k 数据集上,Top-1 准确率相对提升了 14.8%。这意味着模型给出的第一个答案就是正确答案的概率,有了实打实的提高。
最亮眼的是它处理「分布外 (out-of-distribution)」分子的能力。这是检验 AI 模型是不是只会「背书」的试金石。很多模型一碰到自己训练时没见过的全新分子类型,表现就一落千丈。RARB 在这方面表现得非常稳健。原因就在于,即使一个全新的分子整体上是陌生的,但它的某些局部结构很可能在数据库里有先例。检索机制就像一个定海神针,总能给模型找到一个立足于已知化学知识的出发点,避免它天马行空地胡乱猜测。
这个「检索增强」的思路,可能代表了 AI 合成路线规划的一个重要方向。我们正在从训练一个试图「记住」所有化学反应的超级大脑,转向构建一个懂得如何「查阅资料」并进行推理的专家系统。这更接近人类化学家的工作模式。
而且,这个框架的扩展性很好。底层的生成模型是可以替换的。这意味着这套「外脑」系统有潜力嫁接到其他逆合成工具上,让它们也变得更聪明。这让我们看到,未来的药物发现平台或许不再是单一功能的模型,而是一个个懂得协同工作、会查文献、会推理的智能体。
📜Title: Advancing Retrosynthesis with Retrieval-Augmented Graph Generation
🌐Paper: https://ojs.aaai.org/index.php/AAAI/article/download/34203/36358
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-a2mYpWGh-1766808738058)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
4. 聚合物 AI 模型:性能优异,但它真的懂化学吗?
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-J9keL0jq-1766808738059)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
基础模型(Foundation Models)正在席卷各个领域,现在轮到高分子材料科学了。我们都希望 AI 能像一位资深化学家一样,看一眼分子结构就知道它的性质。最近的一项研究朝着这个方向迈出了一步,但其结果也给我们泼了一盆冷水。
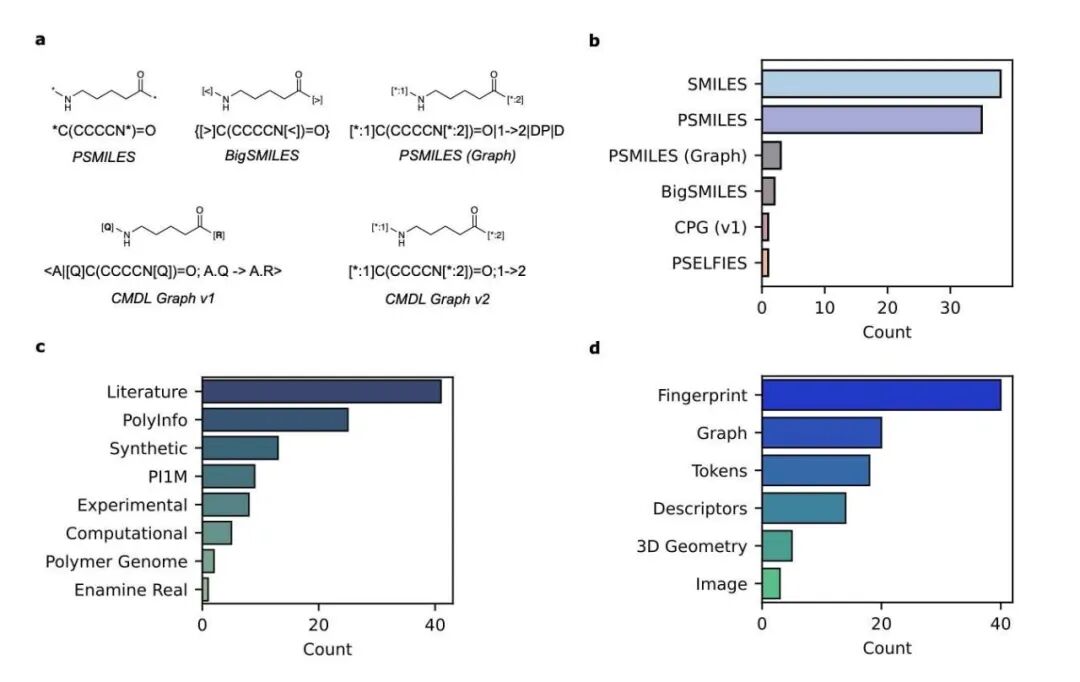
首先,这项工作值得称赞。为聚合物设计一种好的结构表示法是件麻烦事。它不像小分子那样结构固定。聚合物有重复单元、不同的链长、支链和复杂的拓扑结构。研究者们提出了一种基于 SMILES 的新颖图表示法,试图把这些关键特征都编码进去。
基于这种表示法,他们训练了一个名为 SMI-TEDPOLYMER289M 的模型。结果如何?相当不错。在 28 个基准数据集上,这个模型在预测电子、物理、光学和气体阻隔性等多种性能时,都达到了当前最佳或接近最佳的水平。看到这里,你可能会觉得,AI 理解聚合物化学指日可待。
但研究者们没有止步于此,他们做了一件至关重要的事:对照实验。这是这篇论文最精彩的部分。他们故意向模型输入了一些化学上或语义上完全错误的「乱码」SMILES 字符串。
接下来发生的事情,就有点出人意料了。模型在处理这些无效输入时,性能依然很强。
这就引出了一个核心问题。这就像你教一个学生物理,然后给他一道写满了乱码的题目,他却依然给出了正确答案。这时你不会夸他聪明,反而会怀疑他是不是把整本习题册的答案都背下来了,只是根据题目里零星的几个数字或符号,在记忆库里做了个匹配。
研究者通过误差分析和注意力图(attention maps)发现,模型似乎就是这么干的。它并不是在「理解」化学语法,而是在整个序列空间里进行插值。只要输入的字符串在某种程度上与训练数据里的某些序列「长得像」,它就能根据统计规律「猜」出答案,而不管这个字符串在化学上有没有意义。
这告诉我们,至少在目前阶段,这些模型更像是一个见多识广、精于模仿的学徒,而不是一个真正掌握了第一性原理的大师。它的成功很大程度上依赖于训练数据的分布,而不是对化学规则的内在理解。
这篇论文的真正价值,不仅仅是发布了一个高性能模型。它更像一个警示。它提醒我们,在为 AI 模型的性能欢呼时,必须设计严谨的对照实验,去探究模型到底学到了什么。否则,我们很可能会被漂亮的测试分数所蒙蔽。
📜Title: Understanding Structural Representation in Foundation Models for Polymers
🌐Paper: https://arxiv.org/abs/2512.11881
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-QmVWKy8o-1766808738059)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
5. EXAONE Path 2.5:用多组学数据打通病理图像
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-XpuqDtwD-1766808738059)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
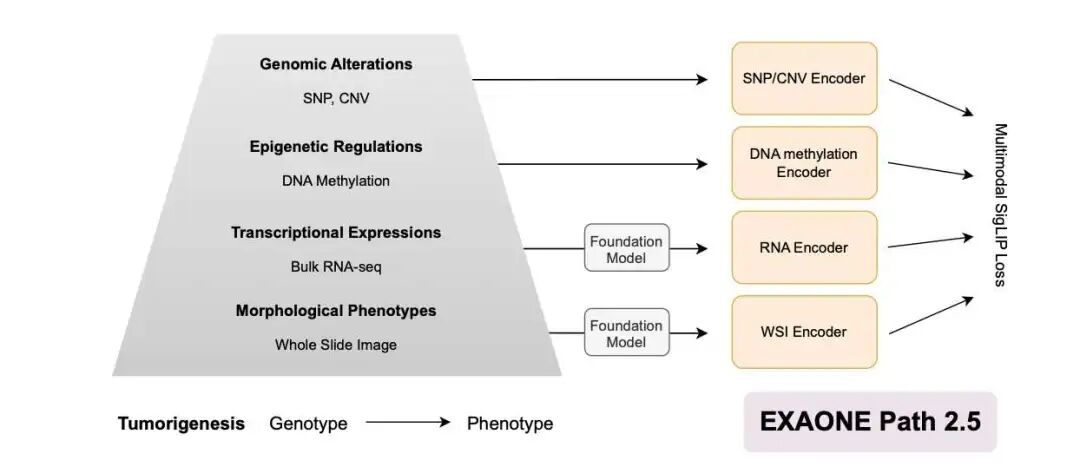
在计算病理学领域,我们手里有海量的全切片图像(Whole-Slide Images, WSI)。这些图像很直观,但它们只讲了故事的一半------也就是肿瘤的「表型」。它长什么样,细胞形态如何。但驱动这一切的「基因型」是什么?这是我们一直想弄清楚的核心问题。
EXAONE Path 2.5 这个模型,就是为了回答这个问题而设计的。
这就像破案。病理切片是犯罪现场的照片,信息量巨大,但光看照片还不够。你还需要 DNA 证据(基因组学)、指纹(表观遗传学)和目击者证词(转录组学)。大多数 AI 模型只擅长分析照片,而 EXAONE Path 2.5 的目标是把所有线索都整合起来。
要把这些完全不同类型的数据对齐,挑战巨大。图像是像素矩阵,RNA-seq 是一堆基因表达计数。这好比让一个画家和一个股票分析师用同一种语言交流,难度可想而知。
研究者们用了几个很方法来解决这个难题。
首先是 多模态 SigLIP 损失函数。传统的对比学习方法,有点像是在搞「拉郎配」,强制让一张图像和它对应的文本配对,同时排斥所有其他选项。当你有五种不同类型的数据时(图像、基因、转录等),这种方法就会出问题。比如,图像和基因的配对,可能会与图像和转录组的配对相互「竞争」,导致模型顾此失彼。SigLIP 的做法是,让每一对正样本(比如,这个图像块和它对应的基因表达谱)自成一派,只跟负样本对比。这样一来,不同模态之间的互补信息就能被更好地保留和编码,而不是在内耗中丢失。
其次是 F-RoPE(Fragment-aware Rotary Positional Encoding)模块。病理切片上的组织不是一盘散沙,它的空间结构至关重要。肿瘤细胞在哪里,基质细胞在哪里,免疫细胞如何浸润,这些位置信息反映了肿瘤的生物学行为。F-RoPE 就像给模型装上了一个高精度 GPS,让它能理解切片上不同区域的空间拓扑关系。这样,模型才能把特定区域的组织学特征,与潜在的分子状态变化精确地联系起来。
这个框架还有一个务实的设计:它为 WSI 和 RNA-seq 数据分别使用了领域专用的基础模型。这相当于先请了两位专家------一位是经验丰富的病理学家,负责解读图像;另一位是顶尖的生物信息学家,负责分析转录数据。有了这些专家提供的稳定、高质量的初始信息(即 embeddings),后续的多模态对齐工作就事半功倍,尤其是在训练数据有限的情况下。
EXAONE Path 2.5 用更少的参数和预训练数据,在包含 80 项任务的 Patho-Bench 基准测试上,表现优于或持平于当前最好的模型。这证明了它的设计效率,说明模型更好地抓住了生物学本质,而不是靠堆砌算力。
经过多模态对齐后,模型学习到的表征空间在生物学上更有意义。比如,不同器官和组织的聚类效果变得更好。这意味着模型真正理解了从基因型到表型的对应关系。
对做药物研发的人来说,这意味着什么?这不仅仅是又一个新算法。它向着构建患者肿瘤的「数字孪生」迈出了一步。我们可以利用这种模型,更精准地筛选生物标志物,或者为临床试验筛选合适的患者。甚至可以想象,未来我们不仅是针对某个蛋白靶点做虚拟筛选,而是针对一个复杂的、包含多组学信息的肿瘤微环境模型进行筛选。这才是这项工作真正令人兴奋的地方。
📜Paper: https://arxiv.org/abs/2512.14019v1
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-ujVROrws-1766808738059)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
6. HeMeNet:一个模型同时搞定蛋白预测六大任务
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-qGJGzK95-1766808738059)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
在药物研发领域,我们经常面临一个现实问题:数据太少,尤其是高质量的标注数据。比如,我们可能有大量关于蛋白稳定性的数据,但对于某个新靶点的亲和力数据却寥寥无几。如果能让模型从一个任务中学到的知识迁移到另一个任务上,就能大大缓解这个问题。这正是多任务学习(Multi-task Learning)的用武之地。
HeMeNet 这个模型就是沿着这个思路设计的。它不是训练一个模型只干一件事,而是让一个模型同时学习六件生物学上相关的事,包括蛋白 - 蛋白亲和力、溶解度、热稳定性等。
它的工作原理是这样的。
首先,模型如何「看」一个蛋白质?传统的模型可能会简化处理,比如只看氨基酸残基的中心碳原子(Cα)。但 HeMeNet 不一样,它构建了一个「异构全原子图」。这意味着模型能看到蛋白质里的每一个原子------碳、氢、氧、氮,以及它们之间的化学键。这种高分辨率的视角至关重要。因为像药物结合这样的精细事件,最终都归结为原子级别的相互作用。只看骨架,会丢失太多关键的化学信息。
其次,蛋白质在三维空间里是自由的,可以任意旋转、移动。它的生物学功能并不会因为你换个角度观察就改变。一个好的计算模型必须尊重这个物理事实。HeMeNet 采用了 E(3) 等变(E(3)-equivariant)架构,这听起来很复杂,但核心思想很简单:你把输入的蛋白质分子在空间里旋转一下,模型的预测结果(比如结合能这个数值)保持不变。这让模型不需要浪费算力去学习所有可能的空间朝向,从一开始就走在正确的物理轨道上。
一个模型怎么同时处理六个不同的任务而不「精神分裂」?研究者设计了一个「任务感知读出机制」。你可以把它想象成模型的核心部分是一个知识渊博的通才,它对蛋白质结构有着深刻、普适的理解。然后,针对每一个具体任务(比如预测亲和力或稳定性),都有一个专门的「专家」模块。这个专家模块会从通才那里获取信息,然后给出针对性的答案。这种设计避免了不同任务在训练过程中互相干扰,确保了每个任务都能达到很好的效果。
实验结果也证明了这一点。在一个新构建的基准测试集上,HeMeNet 在多项任务中,尤其是在亲和力预测上,表现超过了现有的单一任务模型。这说明,让模型「博学多才」确实能帮助它在特定领域做得更专业。对于一线研发人员来说,这意味着未来我们或许可以用更少的数据,训练出更强大、更可靠的预测模型。
📜Title : HeMeNet: Heterogeneous Multichannel Equivariant Network for Protein Multi-task Learning
🌐Paper : https://ojs.aaai.org/index.php/AAAI/article/view/32000/34155
💻Code: https://github.com/hanrthu/HeMeNet
--- 完 ---