BEAUT: Bile acid Enzyme Announcer Unit Tool
https://doi.org/10.1016/j.cell.2025.07.017
GitHub - skystreet8/BEAUT --- GitHub - skystreet8/BEAUT
安装
bash
cd Software
git clone https://github.com/skystreet8/BEAUT
cd BEAUT
conda env create -f environment.yml
conda activate beaut
# 配置环境
mamba install python=3.10 diamond=2.1.9 -c bioconda
mamba install nomkl -y
pip install torch --index-url https://download.pytorch.org/whl/cpu
pip install fair-esm
tar -zxvf nltk_data.tar.gz
cp -r nltk_data/ ~/
#
# 下载模型文件 https://zenodo.org/records/15388149 解压得到data/和data_augmentation/
# 解压后 将 data/ 文件夹放置在 BEAUT/ 目录下
# 将原来有的 data_augmentation_data/ 文件夹放置在 BEAUT/data_augmentation/ 下,然后将 BEAUT/data_augmentation/data_augmentation_data/ 目录重命名为 BEAUT/data_augmentation/data/
# 下载其他文件 https://zenodo.org/records/15388149
# 将 results.tar.gz 文件放入 BEAUT/data_augmentation/PocketMatch/ 目录并解压
运行
BEAUT/esm/esm-extract.sh 修改一下这个文件
bash
#export CUDA_VISIBLE_DEVICES=0
#python scripts/extract.py esm2_t33_650M_UR50D $1 $2 --repr_layers 33 --include mean --toks_per_batch 2048
################### up raw #######################
# 1. 禁用 CUDA 变量(或者直接删掉这一行)
# export CUDA_VISIBLE_DEVICES=0
# 2. 设置 CPU 线程优化(充分利用你的 92 核)
# 建议不要全占满,设置 80 个核心用于计算,留一点给系统
export OMP_NUM_THREADS=80
export MKL_NUM_THREADS=80
# 3. 修改 python 命令
# - 添加 --device cpu (强制使用 CPU)
# - 调大 --toks_per_batch (因为你有 900G 内存,可以显著调大以提升并行度)
python scripts/extract.py esm2_t33_650M_UR50D $1 $2 \
--repr_layers 33 \
--include mean \
--toks_per_batch 16384 \
--nogpu准备 BEAUT/scripts/filter_non_enzymes_3.0.py
python
import re
import argparse
import pandas as pd
from functools import partial
from nltk import word_tokenize
import logging
from utils import *
# Logging Configuration
logging.basicConfig(level=logging.INFO, format='%(levelname)s: %(message)s')
logger = logging.getLogger('PreFilter')
def get_parser():
parser = argparse.ArgumentParser(description='Pre-filter sequences using eggNOG before BEAUT prediction')
parser.add_argument('-f', '--faa', required=True, help='Input protein sequence file (.fasta/.faa)')
parser.add_argument('-e', '--egg', required=True, help='eggNOG annotation file (.annotations)')
parser.add_argument('-o', '--out_prefix', required=True, help='Output file prefix')
return parser
def main():
args = get_parser().parse_args()
# Define eggNOG reader
read_tsv = partial(pd.read_csv, sep='\t', comment='#',
names=['query', 'seed_ortholog', 'evalue', 'score','eggNOG_OGs', 'max_annot_lvl', 'COG_category', 'Description',
'Preferred_name', 'GOs', 'EC', 'KEGG_ko', 'KEGG_Pathway', 'KEGG_Module', 'KEGG_Reaction', 'KEGG_rclass',
'BRITE', 'KEGG_TC', 'CAZy', 'BiGG_Reaction', 'PFAMs'])
# Exclusion keywords (From original BEAUT logic)
filter_ase_words = {'permease', 'peptidase', 'helicase', 'exonuclease', 'atpases', 'heparinase', 'dipeptidyl-peptidase',
'dextransucrase', 'sortase', 'endonuclease', 'aminopeptidase', 'oligopeptidase',
'oligoendopeptidase', 'carboxypeptidase', 'permeases', 'caspase', 'primase', 'atpase',
'pip-5-kinases', "5'-nucleotidase", 'ceramidase', 'metallopeptidase', 'nuclease', 'melibiase',
'trehalase', 'amylase', 'xyloglucanase', 'lectin/glucanases', 'proteinase', 'collagenase',
'ribonuclease', 'calcium-release', 'terminase', 'excisionase', 'metalloprotease', 'polymerase',
'topoisomerase', 'd-transpeptidase', 'depolymerase', 'beta-glucanase', 'elastase',
'alpha-trehalase', 'phospholipase_d-nuclease', 'metallocarboxypeptidase', 'gtpase', 'invertase',
'endonuclease/exonuclease/phosphatase', 'aminopeptidases', 'acylaminoacyl-peptidases', 'telomerase',
'metallo-peptidase', 'replicase', 'fbpase', 'gyrase', 'endo-isopeptidase', 'endoglucanase',
'1,4-beta-xylanase', 'hydrogenlyase', 'carnosinase', 'polygalactosaminidase', 'cam-kinase',
'chitinase', 'chondroitinase', 'metalloproteinase', 'cyclo-malto-dextrinase', 'gamma-secretase',
'metallo-endopeptidase', 'fe-hydrogenase', 'f0f1-atpase', 'trnase', 'l-aminopeptidase', 'dutpase',
'inj_translocase', 'ntpase', 'pectinesterase', 'metallo-endoribonuclease', 'exosortase',
'beta-1,3-glucanase', 'dgtpase', 'carboxypetidase', 'transcriptase',
'protein-phosphocysteine-l-ascorbate-phosphotransferase', 'exoribonuclease', 'coagulase',
'recombinase', 'atcase', 'hydrogenase', 'interferase', 'nadase', 'endosialidase', 'nucleases',
'transposases', 'dipeptidase', 'nickase/helicase', 'rnase', 'dnase/trnase', '-atpase', 'coprotease',
'reactivase', 'metal-chelatase', 'translocase', 'ferrochelatase', 'v-atpase', 'dnase', 'scramblase',
'host-nuclease', 'peptidoglycan-synthase', 'chitosanase', 'peptide-n-glycosidase',
'cobaltochelatase', 'exodeoxyribonuclease', 'disaggregatase', 'integrase', 'relaxase', 'maturase',
'exopeptidases', 'xylanase', 'insulinase', 'metalloendopeptidase', 'arch_atpase', 'de-polymerase',
'transposase', 'proteases', 'dapkinase', 'flippase', 'transpeptidase', 'barnase',
'polygalacturonase', 'n-atpase', 'ld-carboxypeptidase', 'endopeptidase', 'gyrase/topoisomerase',
'autokinase', 'catalase', 'tripeptidases', 'gtpases', 'activase', 'endo-1,4-beta-xylanase',
'amylopullulanase', 'aa_permease', 'metallo-carboxypeptidase', 'chelatase', 'aaa-atpase',
'protease', 'helicases', 'dna-methyltransferase', 'cellulase', 'endoribonuclease', 'levanase',
'excinuclease', 'peptidases', 'convertase', 'apyrase', 'beta-xylanase', 'endodeoxyribonuclease',
'dl-endopeptidase', 'lecithinase', 'resolvase', 'primase/polymerase', 'primase-helicase',
'cutinase', '1,4-beta-cellobiosidase', 'dipeptidylpeptidase', 'd-aminopeptidase',
'amylopullulanase'}
# 1. Load eggNOG Data
logger.info(f'Loading eggNOG annotations from {args.egg}...')
df = read_tsv(args.egg)
# 2. Core Filtering Logic
df_with_ec = df.query('EC != "-"').copy()
df_wo_ec = df.query('EC == "-"').copy()
df_wo_desc = df_wo_ec.query('Description == "-"').copy()
df_with_desc = df_wo_ec.query('Description != "-"').copy()
keep_ase_indexes = []
for t in df_with_desc.itertuples():
desc = t[8].lower()
words = word_tokenize(desc)
if not any([w.endswith('ase') or w.endswith('ases') for w in words]):
continue
for w in words:
if w in filter_ase_words:
break
else:
keep_ase_indexes.append(t[0])
ase_df = df_with_desc.loc[keep_ase_indexes].copy()
# Recover uncharacterized/unknown functions
remove_indexes = [i for i in df_with_desc.index if i not in set(keep_ase_indexes)]
not_ase_df = df_with_desc.loc[remove_indexes].copy()
recover_indexes = [t[0] for t in not_ase_df.itertuples() if any(x in t[8].lower() for x in ['unknown function', 'duf', 'uncharacterised']) or 'non supervised orthologous group' in t[8].lower()]
recovered_df = not_ase_df.loc[recover_indexes].copy()
# Final list of candidate IDs
candidate_df = pd.concat([df_with_ec, ase_df, df_wo_desc, recovered_df], ignore_index=True)
candidate_ids = set(candidate_df['query'].values.tolist())
# 3. Filter FASTA and Save
logger.info(f'Filtering FASTA file: {args.faa}...')
headers, seqs = ReformatFastaFile(args.faa)
filtered_headers = []
filtered_seqs = []
for h, s in zip(headers, seqs):
clean_h = re.split(r'\s+', h)[0]
if clean_h in candidate_ids:
filtered_headers.append(h)
filtered_seqs.append(s)
faa_out = f"{args.out_prefix}_prefiltered.fasta"
SaveFastaFile(faa_out, filtered_headers, filtered_seqs)
logger.info(f'Original count: {len(headers)} | After filtering: {len(filtered_headers)}')
logger.info(f'Filtered FASTA saved to: {faa_out}')
if __name__ == '__main__':
main()我的宏基因组非冗余基因集已经跑过eggnog了
bash
python ${scripts_dir}/filter_non_enzymes_3.0.py \
-f ${ref_protein_file} -e ${eggnog_result}/all_cds_ref_eggnog.emapper.annotations -o ${eggnog_result}/all_cds_ref_pre_BEAUT把结果fasta文件放到 BEAUT/esm 文件夹内,并去除*号
bash
sed '/>/!s/\*//g' ref_BEAUT_prefiltered.fasta > ref_BEAUT_clean.fasta #去除序列的*号使用esm-2进行氨基酸序列预处理
bash
#!/bin/bash
# 1. 路径定义
beaut_root="/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT"
# 建议进入 esm 目录执行,确保相对路径正确
cd ${beaut_root}/esm
# 2. 线程分配
export OMP_NUM_THREADS=20
export MKL_NUM_THREADS=20
export CUDA_VISIBLE_DEVICES=""
# 3. 启动 8 路并行
echo "Starting 8-way parallel extraction..."
for i in {1..8}; do
target_fasta="all_cds_ref_pre_BEAUT_clean.part_00${i}.fasta"
# 每个进程拥有独立的输出目录,避免 I/O 冲突
out_dir="../data/part${i}"
mkdir -p ${out_dir}
# 提交后台任务
nohup bash ${beaut_root}/esm/esm-extract.sh ${target_fasta} ${out_dir} > p${i}.log 2>&1 &
echo "Task ${i} submitted: ${target_fasta} -> ${out_dir}"
done
echo "-------------------------------------------------------"
echo "Check progress: tail -f p1.log"
echo "Monitor CPU: top (Each should be ~2000%)"
echo "-------------------------------------------------------"进行esm-2结果合并
bash
mkdir -p all_embeddings
for i in {1..8}; do
for file in part${i}/embeddings_*.pt; do
# 获取文件名,例如 embeddings_0.pt
fname=$(basename "$file")
# 移动并重命名为 part1_embeddings_0.pt 等
mv "$file" "all_embeddings/part${i}_${fname}"
done
done
cd all_embeddings
count=0
for f in *.pt; do
mv "$f" "embeddings_${count}.pt"
let count++
done
#### 进行预测 ######
cd BEAUT/scripts
python convert_embedding_chunks.py -f all_cds_ref_pre_BEAUT_prefiltered --multiple
python test_bulk.py -f all_cds_ref_pre_BEAUT_prefiltered得到 data/all_cds_ref_pre_BEAUT_prefiltered_results_BEAUT_aug.pkl
构建 BEAUT/scripts/process_bulk_predictions_zp.py
bash
import pandas as pd
import pickle
import os
from utils import ReadFastaFile, SaveFastaFile
# Define paths
result_file = '../data/all_cds_ref_pre_BEAUT_prefiltered_results_BEAUT_aug.pkl'
fasta_input = '../esm/all_cds_ref_pre_BEAUT_clean.fasta'
output_dir = '../data/all_cds_ref_pre_BEAUT_prefiltered_aug/'
# Check if output directory exists
if not os.path.exists(output_dir):
os.makedirs(output_dir)
# Load BEAUT prediction results
print("Loading prediction results...")
with open(result_file, 'rb') as f:
results = pickle.load(f)
# Load original sequences
print("Reading FASTA file...")
headers, seqs = ReadFastaFile(fasta_input)
# Match scores with headers
# Note: results[h][1] is the probability of being an enzyme
print("Matching scores...")
scores = [results[h][1] for h in headers]
# Create DataFrame
df = pd.DataFrame({
'header': headers,
'seq': seqs,
'pred_score': scores
})
# Sort by prediction score descending
df.sort_values('pred_score', ascending=False, inplace=True)
# Filter by threshold 0.5
pos_df = df.query('pred_score >= 0.5').copy()
print(f'{len(pos_df)} sequences were predicted to be positive.')
# Save results in chunks (100,000 sequences per file)
chunk_size = 100000
num_chunks = int(len(pos_df) // chunk_size) + 1
for i in range(1, num_chunks + 1):
start_idx = (i - 1) * chunk_size
end_idx = i * chunk_size
sub_df = pos_df.iloc[start_idx:end_idx]
if sub_df.empty:
continue
# Save CSV
csv_name = f'{output_dir}positive_results_pt{i}.csv'
sub_df.to_csv(csv_name, index=False)
# Save FASTA
fasta_name = f'{output_dir}positive_results_pt{i}.fasta'
sub_headers = sub_df['header'].values.tolist()
sub_seqs = sub_df['seq'].values.tolist()
SaveFastaFile(fasta_name, sub_headers, sub_seqs)
print(f'Saved chunk {i}: {csv_name} and {fasta_name}')
print("All processes completed successfully.")python BEAUT/scripts/process_bulk_predictions_zp.py
得到 data/all_cds_ref_pre_BEAUT_prefiltered_aug/ 里面有 csv和fasta结果信息
长度过滤 按照文献保留 157 到 1074 aa的序列
bash
import pandas as pd
import os
import argparse
def read_fasta(path):
headers, seqs = [], []
with open(path, 'r') as f:
current_seq = []
for line in f:
line = line.strip()
if not line:
continue
if line.startswith('>'):
if current_seq:
seqs.append("".join(current_seq))
headers.append(line[1:])
current_seq = []
else:
current_seq.append(line)
if current_seq:
seqs.append("".join(current_seq))
return headers, seqs
def main():
parser = argparse.ArgumentParser(description='Sync filter for BEAUT CSV and FASTA files.')
parser.add_argument('--input_dir', type=str, required=True, help='Directory with pt1-pt4 files')
parser.add_argument('--min_len', type=int, default=157)
parser.add_argument('--max_len', type=int, default=1074)
args = parser.parse_args()
# 1. Process and Filter CSV Data
csv_files = sorted([f for f in os.listdir(args.input_dir) if f.startswith('positive_results_pt') and f.endswith('.csv')])
print(f"Reading {len(csv_files)} CSV files...")
csv_list = []
for f in csv_files:
temp_df = pd.read_csv(os.path.join(args.input_dir, f))
csv_list.append(temp_df)
full_df = pd.concat(csv_list, ignore_index=True)
full_df['seq_len'] = full_df['seq'].str.len()
# Apply length filter to DataFrame
mask = (full_df['seq_len'] >= args.min_len) & (full_df['seq_len'] <= args.max_len)
filtered_df = full_df[mask].copy()
csv_out = os.path.join(args.input_dir, 'final_enzymes_filtered.csv')
filtered_df.to_csv(csv_out, index=False)
# 2. Process and Filter FASTA Data
fasta_files = sorted([f for f in os.listdir(args.input_dir) if f.startswith('positive_results_pt') and f.endswith('.fasta')])
print(f"Reading and syncing {len(fasta_files)} FASTA files...")
# Use a set for high-speed header lookup
valid_headers = set(filtered_df['header'].values)
fasta_out = os.path.join(args.input_dir, 'final_enzymes_filtered.fasta')
kept_count = 0
with open(fasta_out, 'w') as out_f:
for f in fasta_files:
h_list, s_list = read_fasta(os.path.join(args.input_dir, f))
for h, s in zip(h_list, s_list):
if h in valid_headers:
out_f.write(f">{h}\n{s}\n")
kept_count += 1
# Final Statistics
print("-" * 40)
print("Filtration Results Summary:")
print(f"Total sequences input : {len(full_df)}")
print(f"Sequences kept : {len(filtered_df)}")
print(f"Sequences removed : {len(full_df) - len(filtered_df)}")
print(f"Output CSV : {csv_out}")
print(f"Output FASTA : {fasta_out}")
print("-" * 40)
if __name__ == "__main__":
main()python BEAUT/scripts/length_filter.py python length_filter.py --input_dir data/all_cds_ref_pre_BEAUT_prefiltered_aug/
得到 all_cds_ref_pre_BEAUT_length_filtered.fasta
CLEAN 安装
Enzyme function prediction using contrastive learning | Science
bash
cd ~/software
git clone https://github.com/tttianhao/CLEAN
cd CLEAN/app
conda create -n clean python=3.10 -y
conda activate clean
# 装 PyTorch(CPU 版示例)
mamba install pytorch==1.11.0 torchvision torchaudio cpuonly -c pytorch
# 其余 Python 依赖
pip install -r requirements.txt
# 把 CLEAN 自身也装进环境
python build.py install
# 下载 https://drive.google.com/file/d/1kwYd4VtzYuMvJMWXy6Vks91DSUAOcKpZ/view?usp=sharing
# 复制到data/
cd data
unzip pretrained.zip
# 重命名为解压结果为 pretrained/
cd ..
# 把length_filter.py结果的fasta放到data/inputs
nohup python CLEAN_infer_fasta.py --fasta_data all_cds_ref_pre_BEAUT_length_filtered结果在 CLEAN/app/results/inputs
python
import pandas as pd
from ec_utils import eval_ec, sort_ecs
import os
# --- 1. Load the single CLEAN results file ---
path_clean_maxsep = '/home/zhongpei/hard_disk_sda2/zhongpei/Software/CLEAN/app/results/inputs/all_cds_ref_pre_BEAUT_length_filtered_maxsep.csv'
# CLEAN output has 6 columns: Header + 5 Predictions
merged_ec_df = pd.read_csv(
path_clean_maxsep,
names=['header', 'pred_1', 'pred_2', 'pred_3', 'pred_4', 'pred_5']
)
merged_ec_df.fillna('-', inplace=True)
# Apply eval_ec to each prediction column
for i in range(1, 6):
merged_ec_df[f'pred_{i}'] = merged_ec_df[f'pred_{i}'].apply(eval_ec)
# Create a dictionary for fast mapping: {header: sorted_ec_list}
# range(2, 7) corresponds to t[2] through t[6] in itertuples (pred_1 to pred_5)
ec_predictions = {
t[1]: sort_ecs([t[i][0] for i in range(2, 7) if t[i][0] != 'NONE'])
for t in merged_ec_df.itertuples()
}
# --- 2. Load and merge the 4 metadata parts (pt1 to pt4) ---
metadata_dfs = []
metadata_dir = '../data/all_cds_ref_pre_BEAUT_prefiltered_aug'
for i in range(1, 5):
# Construct the path for positive_results_pt1.csv, pt2.csv, etc.
pt_path = os.path.join(metadata_dir, f'positive_results_pt{i}.csv')
if os.path.exists(pt_path):
pt_df = pd.read_csv(pt_path)
metadata_dfs.append(pt_df)
else:
print(f"Warning: {pt_path} not found.")
merged_df = pd.concat(metadata_dfs, ignore_index=True)
# --- 3. Assign CLEAN ECs to the merged metadata ---
# t[1] is the header/ID in your metadata csv
merged_df = merged_df.assign(
clean_ec=[ec_predictions.get(t[1], []) for t in merged_df.itertuples()]
)
# --- 4. Save final output ---
output_final = '../data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_length_filtered_EC.csv'
# Ensure the output directory exists
os.makedirs(os.path.dirname(output_final), exist_ok=True)
merged_df.to_csv(output_final, index=False)
print(f"Merge completed successfully.")
print(f"Total sequences in metadata: {len(merged_df)}")
print(f"Total sequences with CLEAN annotations: {len(ec_predictions)}")
print(f"Final file saved to: {output_final}")python add_clean_predictions_zp.py
得到 all_cds_ref_pre_BEAUT_length_filtered_EC.csv
python
import pickle
from ec_utils import sort_ecs_short, shorten_ec
import pandas as pd
from collections import Counter
import ast
def shorten_ec_list(t):
return [shorten_ec(s) for s in t]
# 1. Load the KEGG name dictionary
ec2names = pickle.load(open('../data/kegg_ec2names.pkl', 'rb'))
# 2. Read your 4-column CSV file
input_path = '/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_length_filtered_EC.csv'
raw_df_aug = pd.read_csv(input_path)
# 3. Safely convert string representation of list to Python list
# It uses ast.literal_eval which is safer than eval()
raw_df_aug['clean_ec_short'] = raw_df_aug['clean_ec'].apply(
lambda x: ast.literal_eval(x) if isinstance(x, str) else x
)
# 4. Correct outdated EC numbers
for i in raw_df_aug.index:
short_ecs = raw_df_aug.at[i, 'clean_ec_short']
if not isinstance(short_ecs, list):
continue
for j in range(len(short_ecs)):
if short_ecs[j] == '3.3.1.1':
short_ecs[j] = '3.13.2.1'
elif short_ecs[j] == '3.3.1.2':
short_ecs[j] = '3.13.2.2'
elif short_ecs[j] == '3.3.1.3':
short_ecs[j] = '3.2.1.148'
# 5. Collapse to 3-digit categories (e.g., 3.5.1.24 -> 3.5.1)
raw_df_aug['clean_ec_short'] = raw_df_aug['clean_ec_short'].apply(shorten_ec_list)
# 6. Optimized counting (replaces the slow nested loop and t[26])
print("Calculating global EC statistics...")
global_counter = Counter()
for ecs in raw_df_aug['clean_ec_short']:
global_counter.update(ecs)
# Get sorted unique EC list
all_ecs = sort_ecs_short(list(global_counter.keys()))
# 7. Generate names from the dictionary
names = []
for ec in all_ecs:
try:
digits = ec.split('.')
ec1 = digits[0]
ec2 = '.'.join(digits[:2])
# Fetching names with "Unknown" as fallback
n1 = ec2names.get(ec1, "Unknown")
n2 = ec2names.get(ec2, "Unknown")
n3 = ec2names.get(ec, "Unknown")
names.append(', '.join([n1, n2, n3]))
except:
names.append("Name mapping not found")
# 8. Create summary DataFrame and save
ec_stat_df_aug = pd.DataFrame({
'EC': all_ecs,
'total': [global_counter[ec] for ec in all_ecs],
'name': names
})
output_path = '/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_length_filtered_EC_stat.csv'
ec_stat_df_aug.to_csv(output_path, index=False)
print(f"Success! Statistics saved to {output_path}")python ec_stat_zp.py
得到 all_cds_ref_pre_BEAUT_length_filtered_EC_stat.csv
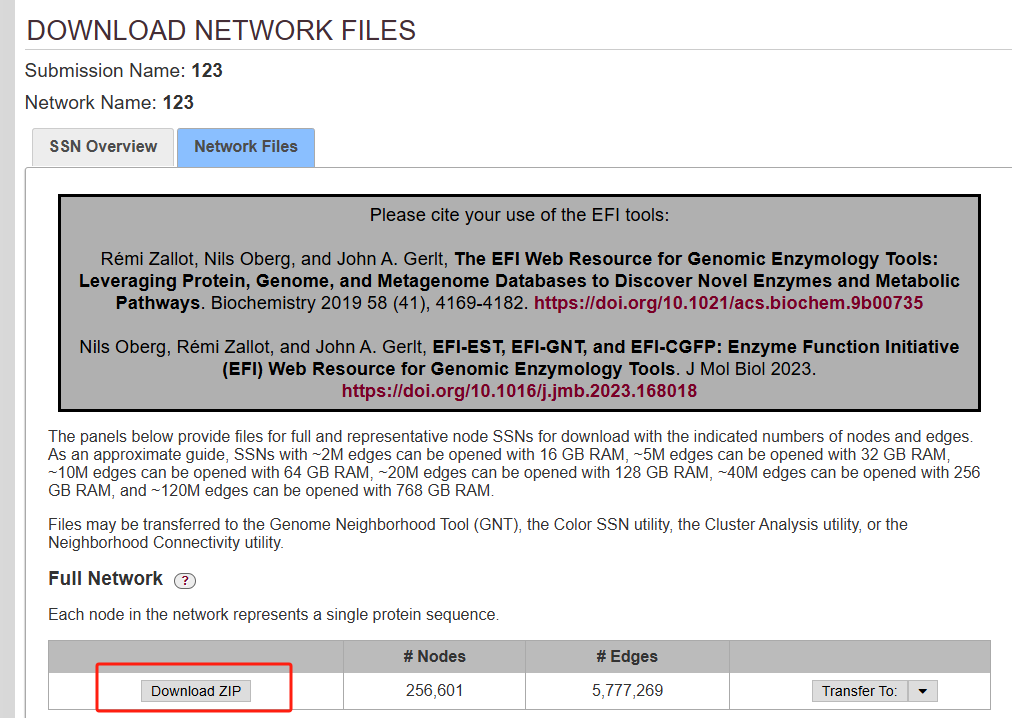
使用EFI-EST获取蛋白聚类
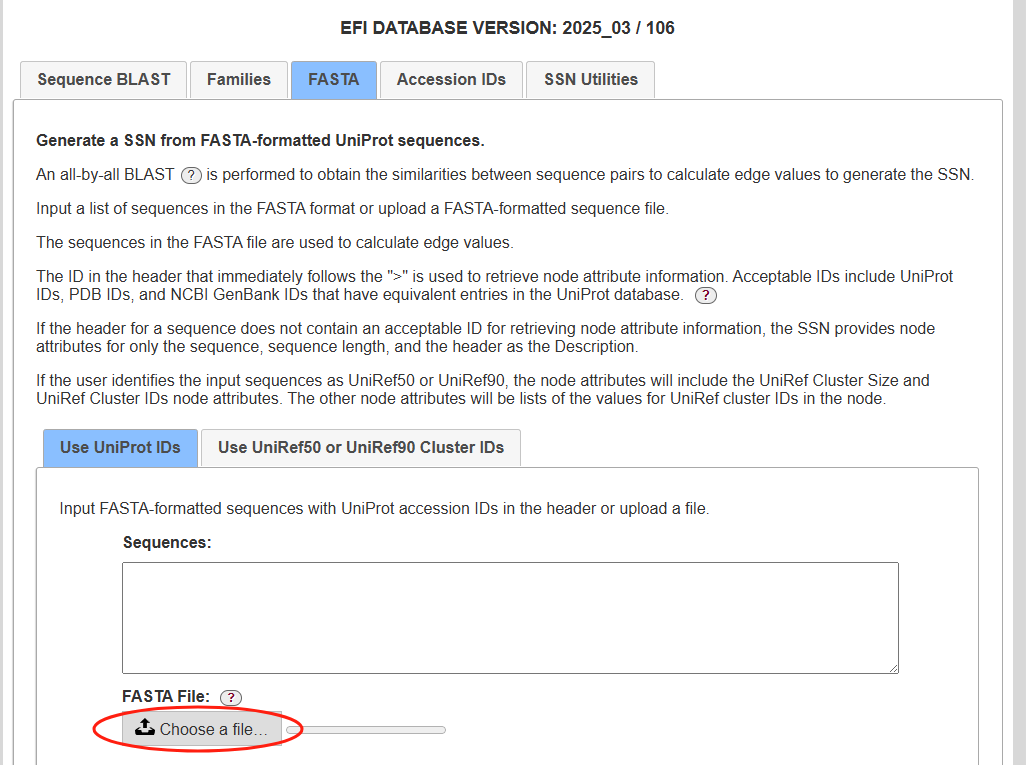
结果邮件会出来
python
import pickle
import networkx as nx
from copy import deepcopy
import xml.parsers.expat
import time
import logging
logger = logging.getLogger('process_xgmml_graph')
logger.addHandler(logging.StreamHandler())
logger.setLevel(logging.INFO)
class XGMMLParserHelper(object):
def __init__(self):
self._graph = nx.DiGraph()
self._parser = xml.parsers.expat.ParserCreate()
self._parser.StartElementHandler = self._start_element
self._parser.EndElementHandler = self._end_element
self._tagstack = list()
self._network_att_el = dict()
self._current_att_el = dict()
self._current_list_att_el = list()
self._current_obj = dict()
def _start_element(self, tag, attr):
self._tagstack.append(tag)
if tag == 'graph':
self._network_att_el = dict()
if tag == 'node' or tag == 'edge':
self._current_obj = dict(attr)
if tag == 'att' and (self._tagstack[-2] == 'node' or
self._tagstack[-2] == 'edge'):
if 'value' in attr:
self._current_att_el = self._parse_att_el(self._current_att_el,
tag, attr)
elif attr['type'] == 'list':
self._current_list_name = attr['name']
self._current_att_el[attr['name']] = list()
if tag == 'att' and (self._tagstack[-2] == 'att'):
self._current_list_att_el = dict(attr)
if 'value' in attr:
self._current_list_att_el = self._parse_att_el(
self._current_list_att_el, tag, attr)
self._current_att_el[self._current_list_name].append(
self._current_list_att_el[attr['name']])
if tag == 'att' and self._tagstack[-2] == 'graph':
if 'value' in attr:
self._network_att_el[attr['name']] = attr['value']
def _parse_att_el(self, att_el, tag, attr):
if 'value' in attr:
if attr['type'] == 'string':
att_el[attr['name']] = attr['value']
elif attr['type'] == 'real':
att_el[attr['name']] = float(attr['value'])
elif attr['type'] == 'integer':
att_el[attr['name']] = int(attr['value'])
elif attr['type'] == 'boolean':
att_el[attr['name']] = bool(attr['value'])
else:
raise NotImplementedError(attr['type'])
return att_el
def _end_element(self, tag):
if tag == 'node':
for k in remove_node_keys:
if k in self._current_att_el:
del self._current_att_el[k]
for k in keep_node_keys_repl:
if k in self._current_att_el:
self._current_att_el[keep_node_keys_repl[k]] = deepcopy(self._current_att_el[k])
del self._current_att_el[k]
if 'label' in self._current_obj:
if 'label' in self._current_att_el:
self._current_att_el['@label'] = self._current_att_el['label']
del self._current_att_el['label']
self._graph.add_node(self._current_obj['id'],
label=self._current_obj['label'],
**self._current_att_el)
else:
self._graph.add_node(self._current_obj['id'],
**self._current_att_el)
self._current_att_el = dict()
elif tag == 'edge':
self._current_att_el['fident'] = self._current_att_el['%id']
del self._current_att_el['%id']
self._graph.add_edge(self._current_obj['source'],
self._current_obj['target'],
**self._current_att_el)
self._current_att_el = dict()
self._tagstack.pop()
def parseFile(self, file):
self._parser.ParseFile(file)
def graph(self):
return self._graph
def graph_attributes(self):
return self._network_att_el
def XGMMLReader(graph_file):
parser = XGMMLParserHelper()
parser.parseFile(graph_file)
return parser.graph()
ssn_name = 'alnscore60_full'
keep_node_keys_repl = {
'Taxonomy ID': 'taxonomy_id',
'UniProt Annotation Status': 'uniprot_annotation_status',
'SwissProt Description': 'swissprot_description',
'Sequence Length': 'length',
'Sequence Status': 'sequence_status',
'Number of IDs in Rep Node': 'num_members'
}
remove_node_keys = ['Sequence Source', 'Other IDs', 'Gene Name', 'NCBI IDs', 'List of IDs in Rep Node', 'Superkingdom', 'Kingdom', 'Phylum', 'Class',
'Order', 'Family',
'Genus', 'Species', 'PDB', 'TIGRFAMs', 'InterPro (Domain)', 'InterPro (Family)', 'InterPro (Homologous Superfamily)',
'InterPro (Other)', 'BRENDA ID', 'Cazy Name', 'GO Term', 'KEGG ID', 'PATRIC ID', 'STRING ID', 'HMP Body Site', 'HMP Oxygen',
'P01 gDNA', 'Rhea', 'AlphaFold', 'Sequence']
start_time = time.monotonic()
graph = XGMMLReader(
open(f'/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_full_ssn.xgmml',
'rb')
)
end_time = time.monotonic()
logger.info(f'{round(end_time - start_time, 0)}s used for loading the graph.')
start_time = time.monotonic()
graph = graph.to_undirected()
end_time = time.monotonic()
logger.info(f'{round(end_time - start_time, 0)}s used for transforming the graph to undirected.')
logger.info(f'There are {graph.number_of_nodes()} nodes in the graph.')
logger.info(f'There are {graph.number_of_edges()} egdes in the graph.')
n_connected_components = len(list(nx.connected_components(graph)))
logger.info(f'There are {n_connected_components} clusters in the graph.')
cluster_sizes = [len(c) for c in nx.connected_components(graph)]
logger.info(f'Largest cluster: {max(cluster_sizes)}')
logger.info(f'There are {sum([n >= 3 for n in cluster_sizes])} clusters with >= 3 sequences.')
logger.info(f'There are {sum([n >= 5 for n in cluster_sizes])} clusters with >= 5 sequences.')
logger.info(f'There are {sum([n >= 10 for n in cluster_sizes])} clusters with >= 10 sequences.')
logger.info(f'There are {sum([n >= 50 for n in cluster_sizes])} clusters with >= 50 sequences.')
logger.info(f'There are {sum([n >= 100 for n in cluster_sizes])} clusters with >= 100 sequences.')
logger.info(f'There are {sum([n >= 500 for n in cluster_sizes])} clusters with >= 500 sequences.')
logger.info(f'There are {sum([n >= 1000 for n in cluster_sizes])} clusters with >= 1000 sequences.')
logger.info(f'There are {sum([n == 1 for n in cluster_sizes])} singletons.')
conn_comps = list(nx.connected_components(graph))
conn_comps.sort(key=lambda t: len(t), reverse=True)
conn_comps_to_save = []
for comp in conn_comps:
conn_comps_to_save.append([])
for n in comp:
conn_comps_to_save[-1].extend(graph.nodes[n]['Description'])
with open(f'/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_prefiltered_{ssn_name}_clusters.pkl', 'wb') as f:
pickle.dump(conn_comps_to_save, f)
f.close()python process_xgmml_graph_zp.py
得到 all_cds_ref_pre_BEAUT_prefiltered_alnscore60_full_clusters.pkl
python
import pickle
import pandas as pd
from ec_utils import shorten_ec
from collections import defaultdict
df = pd.read_csv('/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_length_filtered_EC.csv')
df.set_index('header', inplace=True)
clu_60 = pickle.load(open('/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/all_cds_ref_pre_BEAUT_prefiltered_alnscore60_full_clusters.pkl', 'rb'))
cluster_info = pd.DataFrame({'cluster_id':
[f'cluster_{i}' for i in range(1, len(clu_60) + 1)],
'size':
[len(t) for t in clu_60]
})
cluster_ec_info = []
for indexes in clu_60:
qdf = df.loc[indexes].copy().sort_values('pred_score', ascending=False)
ec_dict = defaultdict(int)
for t in qdf.itertuples():
this_ecs = [shorten_ec(s) for s in eval(t[-1])]
this_ecs = set(this_ecs)
for k in this_ecs:
ec_dict[k] += 1
ec_info = ''
most_common_ecs = list(ec_dict.items())
most_common_ecs.sort(key=lambda x: x[1], reverse=True)
most_common_ecs = most_common_ecs[:3]
for t in most_common_ecs:
ec_info += f'EC:{t[0]}, {round(t[1] / len(qdf) * 100, 1)}%; '
cluster_ec_info.append(ec_info[:-2])
top1_ec = []
top1_ec_ratio = []
for s in cluster_ec_info:
if s == 'NONE':
top1_ec.append('-')
top1_ec_ratio.append('-')
else:
try:
top1_ec.append(s.split('; ')[0].split(',')[0])
top1_ec_ratio.append(s.split('; ')[0].split(', ')[1])
except:
print(s)
raise
top2_ec = []
top2_ec_ratio = []
for s in cluster_ec_info:
if s == 'NONE':
top2_ec.append('-')
top2_ec_ratio.append('-')
else:
strings = s.split('; ')
if len(strings) == 1:
top2_ec.append('-')
top2_ec_ratio.append('-')
else:
top2_ec.append(strings[1].split(', ')[0])
top2_ec_ratio.append(strings[1].split(', ')[1])
top3_ec = []
top3_ec_ratio = []
for s in cluster_ec_info:
if s == 'NONE':
top3_ec.append('-')
top3_ec_ratio.append('-')
else:
strings = s.split('; ')
if len(strings) < 3:
top3_ec.append('-')
top3_ec_ratio.append('-')
else:
top3_ec.append(strings[2].split(', ')[0])
top3_ec_ratio.append(strings[2].split(', ')[1])
cluster_info = cluster_info.assign(top1_ec=top1_ec, top1_ec_ratio=top1_ec_ratio,
top2_ec=top2_ec, top2_ec_ratio=top2_ec_ratio, top3_ec=top3_ec, top3_ec_ratio=top3_ec_ratio)
cluster_info.to_csv('/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/cluster_info.csv',
index=False)
header2clu = {}
for i, t in enumerate(clu_60):
for h in t:
header2clu[h] = i + 1
headers = []
for t in clu_60:
headers.extend(t)
qdf = df.loc[headers].copy()
qdf['cluster'] = [header2clu[t[0]] for t in qdf.itertuples()]
qdf.sort_values(by=['cluster', 'pred_score'], ascending=[True, False], inplace=True)
qdf['cluster'] = qdf['cluster'].astype(str)
for i in qdf.index:
qdf.at[i, 'cluster'] = f"cluster_{qdf.at[i, 'cluster']}"
cols = qdf.columns.values.tolist()
qdf = qdf[cols[:2] + ['cluster'] + cols[2:-1]]
qdf.reset_index(inplace=True)
qdf.to_csv('/home/zhongpei/hard_disk_sda2/zhongpei/Software/BEAUT/data/all_cds_ref_pre_BEAUT_prefiltered_aug/cluster_results.csv',
index=False)python process_ssn_clusters_zp.py
得到 cluster_results.csv 和 cluster_info.csv