
Gap-less and haplotype-resolved genomes of two Hippophae rhamnoides subspecies: Hippophae rhamnoides subsp. mongolica and Hippophae rhamnoides subsp. Sinensis
沙棘(Hippophae rhamnoides ,2n =2x =24)为胡颓子科沙棘属植物,原产于亚洲及欧洲西北部地区。该物种具有极强的抗逆性,可耐受 - 40℃至 40℃的极端温度,同时对干旱与水涝环境均具备良好适应性,因此极易开展人工栽培。沙棘果实富含多种营养物质与生物活性成分,其中黄酮类化合物与维生素 C(VC)含量尤为丰富,这也是其被广泛引种并实现全球栽培的重要原因。近年来,测序技术与基因组组装方法的持续革新,已助力科研人员完成多个物种的染色体级基因组解析,部分物种的基因组组装甚至达到了无缺口的精细标准 1。值得注意的是,目前已有多项研究完成了沙棘属植物的染色体级基因组组装,涉及物种包括蒙古沙棘(H. rhamnoides subsp. Mongolia )2、西藏沙棘 3、中国沙棘 4 以及杂交沙棘(蒙古沙棘 × 中国沙棘)5。然而,这些组装版本的基因组中仍存在大量缺口序列与未锚定的重叠群,这对后续的数据分析与功能研究造成了极大阻碍。
本研究完成了两个代表性沙棘亚种的无缺口单倍型基因组组装 ,其中蒙古沙棘的突出特性为果实大、产量高、含油量高且棘刺稀疏;中国沙棘则表现出抗逆性强、生长速度快及维生素 C 含量高的优势。针对蒙古沙棘,本研究共产生 37 Gb 的 PacBio HiFi 测序数据(测序深度 30×)、67 Gb 的牛津纳米孔(ONT)测序数据(测序深度 54×)以及 73 Gb 的 Hi-C 测序数据。对于中国沙棘,研究团队采用了相似的测序策略,获得 37 Gb 的 PacBio HiFi 测序数据(测序深度 35×)、61 Gb 的 ONT 测序数据(测序深度 60×)和 83 Gb 的 Hi-C 测序数据。本研究利用 hifiasm(v0.19.8-r602)软件,将 PacBio HiFi 与 ONT 测序数据联合组装为基因组重叠群;随后通过 Juicer(v1.6)软件将 Hi-C 测序数据比对至单倍型基因组组装结果,并利用 3D-DNA(v180419)软件完成染色体水平的挂载;借助 Juicebox(v1.11.08)软件对染色体边界及组装错误区域进行人工核查与校正;基于 HiFi 测序数据,利用 quarTeT(v1.2.5)软件填补基因组中的缺口序列。针对比对到染色体末端的 HiFi 测序数据,本研究对其进行重新组装以延长染色体长度,力求获得完整的端粒序列。此外,研究人员利用 GetOrganelle(v1.7.7.1)软件完成了沙棘叶绿体与线粒体基因组的组装。
去除冗余序列后,本研究成功组装获得两个亚种的 12 对染色体,以及叶绿体和线粒体基因组。其中,蒙古沙棘基因组仅含 4 个缺口,中国沙棘基因组含 2 个缺口,分别组装得到 38 个和 42 个端粒序列(图 1A-B)。蒙古沙棘和中国沙棘的单倍型基因组大小分别为 2.33 Gb 和 2.09 Gb,重叠群 N50 值(contig N50)分别达到 97 Mb 和 81 Mb。蒙古沙棘的 PacBio HiFi 测序数据回比效率为 99.55%,且 99.76% 的基因组区域覆盖深度≥5×;BUSCO(v5.8.2)基因组完整性评估结果显示,蒙古沙棘和中国沙棘的完整 BUSCO 基因占比分别为 99.0% 和 98.8%。两个基因组的 Hi-C 互作图谱均呈现清晰的对角线区块,无明显错连现象,表明染色体水平组装准确性高。上述结果证实,本研究已获得两个无缺口的单倍型解析基因组。此外,本研究通过 K-mer 频谱分析、开关错误定量评估等多种互补方法验证组装相位质量:K-mer 分析证实组装结果准确,单倍型间序列差异清晰可辨,无显著污染或嵌合现象;蒙古沙棘和中国沙棘的开关错误率(switch-error rate)分别为 0.8% 和 0.6%,均远低于高质量单倍型组装公认的 1% 阈值。与已报道的沙棘基因组相比,本研究组装版本的连续性显著提升,contig N50 值增加超过 20 倍,支架序列(scaffold)数量大幅减少(图 1C)。
本研究采用 EDTA(v1.9.9)联合 RepeatMasker(v4.1.8)软件进行重复序列注释,结果显示蒙古沙棘和中国沙棘基因组中重复序列占比分别为 71.09% 和 71.96%。两个基因组的重复序列均以长末端重复序列(LTR)为主,分别占蒙古沙棘基因组的 44.74% 和中国沙棘基因组的 46.58%。在 LTR 序列中,Gypsy 类元件为主要组成部分,在蒙古沙棘和中国沙棘中占比分别为 23.44% 和 12.56%。二者 Gypsy 类元件占比的显著差异,主要源于未分类 LTR 序列的比例不同(蒙古沙棘 10.67% vs 中国沙棘 22.82%)。
基于同源序列证据、转录组证据及从头预测(ab initio),本研究分别在蒙古沙棘和中国沙棘基因组中注释到 56,217 个和 57,160 个蛋白质编码基因。除蛋白质编码基因外,蒙古沙棘基因组还包含 791 个 rRNA 基因、1,460 个 tRNA 基因和 4,897 个非编码 RNA(ncRNA)基因;中国沙棘基因组则注释到 1,885 个 rRNA 基因、1,200 个 tRNA 基因和 5,319 个 ncRNA 基因。BUSCO 蛋白质编码基因集完整性评估结果显示,蒙古沙棘和中国沙棘的完整 BUSCO 基因占比分别为 99.2% 和 99.0%;功能注释结果表明,98.05% 的蒙古沙棘蛋白质和 98.15% 的中国沙棘蛋白质可被赋予推测功能。
本研究组装的每个单倍型基因组大小,约为先前发表的近缘亚种染色体级基因组的 1.5 倍(图 1C)。为探究这些额外序列的来源,本研究利用 SyRI(v1.6)软件,将中国沙棘参考基因组 4 与蒙古沙棘、中国沙棘的所有单倍型进行全基因组比对。结果显示,12 条染色体均存在广泛的结构重排,主要表现为频繁的染色体间易位和染色体内倒位(图 1D)。为进一步验证 SyRI 鉴定的倒位事件真实性,本研究分析了中国沙棘两个单倍型的 Hi-C 互作模式,发现对角线外的互作信号与预测的倒位事件完全吻合(图 1E)。与已发表的沙棘基因组 2,4 相比,本研究组装的每个单倍型均包含大量新增序列。为验证这些在早期组装版本 4,5 中缺失的序列是真实存在的基因组片段,而非测序或组装错误导致的假阳性结果,本研究将公开的中国沙棘 HiFi 测序数据 4 回比至本研究组装基因组,发现这些新增区域无测序数据支持;而本研究自身的 HiFi 数据则完全覆盖这些区域,证实其为真实的基因组差异(图 1F)。比对区域中超过 40% 存在非共线性关系,仅倒位事件就影响每个单倍型 147-210 Mb 的序列;此外,非共线性或未比对区域占每个查询基因组的 28.3%-48.4%,表明沙棘属植物存在显著的单倍型特异性变异、种间分化,以及重复序列或结构复杂区域的扩张。例如,中国沙棘单倍型 B(hapB)含 208 Mb 中国沙棘参考基因组中缺失的序列,而蒙古沙棘单倍型 A(hapA)的此类特有序列高达 426 Mb(图 1G)。
为探究这些大规模结构变异的潜在驱动因素,本研究进一步分析了转座子(TE)的贡献。全基因组富集分析显示,LTR/Gypsy 元件的分布并非随机:与随机模拟结果相比,结构重排区域中 LTR/Gypsy 元件显著富集(图 1H)。值得注意的是,染色体断裂点处的 LTR/Gypsy 元件富集程度(hapA:2,360 个;hapB:1,810 个)显著高于整个倒位区域(hapA:1,547 个;hapB:1,308 个)。这些结果表明,LTR/Gypsy 元件通过主动介导染色体断裂与重排,在塑造蒙古沙棘和中国沙棘独特的基因组结构中发挥了关键作用。
两个沙棘亚种在果肉维生素 C(VC)含量和种子含油量方面存在显著差异(图 1I-J):中国沙棘果肉 VC 含量显著高于蒙古沙棘(图 1I)。为阐明该性状的分子机制,本研究分析了抗坏血酸(VC)生物合成途径,发现 GDP-L - 半乳糖磷酸化酶(VTC,基因 ID:Hirsi04aG0052800)、L - 半乳糖脱氢酶(L-GalDH,基因 ID:Hirsi03aG0325000)和 L - 半乳糖酸 - 1,4 - 内酯脱氢酶(GLDase,基因 ID:Hirsi04aG0123700)等关键基因在中国沙棘中的表达水平显著高于蒙古沙棘(图 1K),与 VC 含量差异趋势一致。统计分析表明,这些基因在两个亚种间的表达差异具有统计学意义(t 检验,p < 0.05),提示其上调表达可能是中国沙棘 VC 积累量更高的重要原因(图 1I)。
与之相反,蒙古沙棘的种子含油量显著高于中国沙棘(图 1J)。三酰甘油(TAG)和脂肪酸生物合成途径的转录组分析显示,蒙古沙棘中 TAG 合成关键酶 ------ 酰基辅酶 A:二酰甘油酰基转移酶(基因 ID:Hirmo06aG0056400),以及一个亚种特异性的甘油 - 3 - 磷酸 O - 酰基转移酶(GPAT,基因 ID:Hirmo09aG0163800)的表达水平显著升高(图 1L);此外,脂肪酸途径中的两个脂肪醛脱氢酶(ALDH,基因 ID:Hirmo07aG0056100 和 Hirmo05aG0138100),以及一个亚种特异性 ALDH 基因(基因 ID:Hirmo05aG0120300)也呈上调表达(图 1M)。统计检验证实,这些基因在蒙古沙棘中的表达量显著高于中国沙棘(t 检验,p < 0.05),支持其对种子含油量提升的贡献(图 1J)。
综上所述,本研究成功组装了蒙古沙棘和中国沙棘的无缺口单倍型解析基因组,并提供了与关键果实品质性状相关的组织特异性转录组数据。这些资源不仅为解析沙棘属植物的基因组结构与基因组成提供了高分辨率参考,还揭示了 VC 和种子油脂亚种特异性积累的分子机制,为沙棘的进化研究与靶向育种奠定了重要基础。
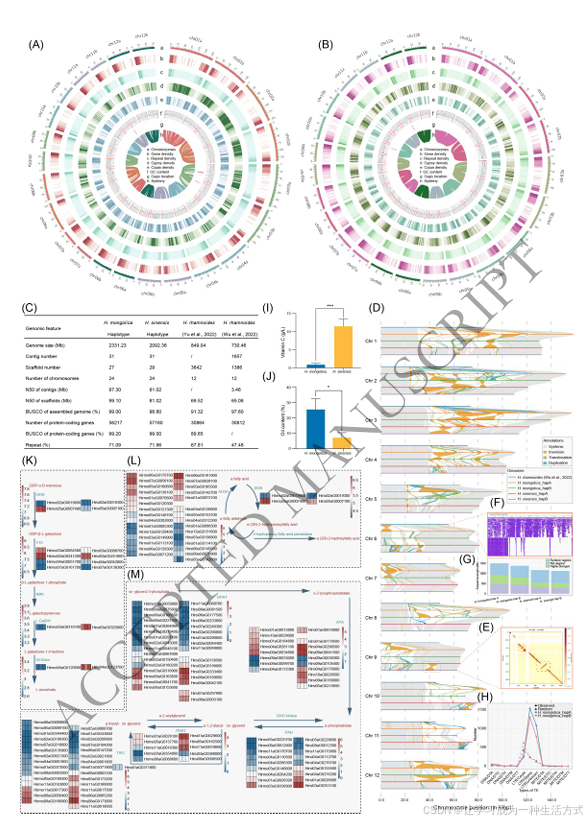
图 1 蒙古沙棘与中国沙棘的基因组及转录组图谱
(A-B)蒙古沙棘(A)与中国沙棘(B)基因组特征的环形图谱 。从外环至内环依次为:(a)染色体;(b)基因密度;(c)重复序列密度;(d)Gypsy 类反转录转座子密度;(e)Copia 类反转录转座子密度;(f)GC 含量,染色体末端的红色线条代表已完成组装的端粒序列;(g)缺口位置;(h)共线性关系。除染色体轨道外,其余轨道颜色越深表示对应特征的密度越高。在共线性轨道中,线条代表同一物种内单倍型 A 与单倍型 B 同源区域间的共线性关系,且仅展示长度超过 100 kb 的比对区域。
(C)本研究新测序的蒙古沙棘与中国沙棘基因组,与已发表参考基因组的组装指标对比 。
(D)5 个沙棘属单倍型解析基因组的共线性关系及结构变异分析。利用 SyRI 软件进行共线性分析,通过两两比对单倍型解析基因组,鉴定各类结构变异。横轴代表 12 条参考染色体(chr1--chr12),单位为 Mb。每条轨道展示查询基因组与参考基因组的比对结果,不同颜色代表不同结构变异类型:灰色为共线性区域,橙色为倒位区域,绿色为易位区域,蓝色为重复区域。
(E)中国沙棘 9 号染色体两个单倍型(chr9a 与 chr9b)间的Hi-C 互作热图 。对角线代表各单倍型内部的互作关系,红色表示互作频率高,反映该区域在三维空间中物理距离较近。
(F)蒙古沙棘 6 号染色体单倍型 A(chr06a,8,617,048--9,607,932 bp)的HiFi 测序序列比对可视化图谱 (IGV)。上图:中国沙棘参考基因组 4 的 HiFi 序列比对至蒙古沙棘单倍型 A 组装序列的结果;下图:蒙古沙棘自身 HiFi 序列比对至其单倍型 A 组装序列的结果。
(G)基于 SyRI 分析统计结果绘制的变异类型堆叠柱状图 。
(H)蒙古沙棘两个单倍型中,与转座子(TE)重叠的倒位断裂点数量统计(每个断裂点长度为 150 bp)。实线代表实际检测到的分布模式,虚线代表随机模拟模式(1000 次随机重排);蓝色与红色分别代表单倍型 A(HapA)与单倍型 B(HapB)。
(I)蒙古沙棘与中国沙棘果肉维生素 C(VC)含量对比 。
(J)蒙古沙棘与中国沙棘种子含油量对比 。数据以平均值 ± 标准差表示(n=3);采用学生 t 检验分析差异显著性(P < 0.05)。
(K-M)关键通路基因的转录本丰度热图 (以 log₂(TPM) 值表示)。其中,(K)抗坏血酸(VC)生物合成通路基因(筛选条件:log₂(TPM) ≥ 1);(L)三酰甘油(TAG)生物合成通路基因;(M)脂肪酸代谢通路基因。基因 ID 以 "Hirmo" 开头代表蒙古沙棘基因,以 "Hirsi" 开头代表中国沙棘基因。每行对应一对直系同源基因,或仅在一个亚种中存在、在另一亚种中无同源序列的亚种特异性基因。仅展示至少在一个样本中 TPM ≥ 1 的基因。