The near-complete genome assembly of Ampelopsis grossedentata provides insights into its origin, evolution, and the regulation of flavonoid biosynthesis
显齿蛇葡萄染色体级别的基因组精细图谱解析------ 揭示物种起源演化及黄酮类化合物生物合成调控机制

显齿蛇葡萄原产于中国南方,因其显著的药用价值与营养价值备受关注;该植物富含二氢杨梅素,故而被誉为 "黄酮之王"。其干燥的茎、叶及嫩梢被称为 "藤茶",既可作为保健饮品日常饮用,也可作为传统药材用于治疗感冒与发热症状。本研究完成了显齿蛇葡萄近乎完整的参考基因组组装,基因组大小为 555.42 Mb;借助 Hi-C 技术辅助挂载,成功将其 20 条染色体中的 18 条组装为无缺口序列。最终获得的基因组挂载于 20 条染色体上,包含 44 个 contig(N50 = 21.93 Mb)与 28 个 scaffold(N50 = 30.45 Mb),共注释得到 25999 个蛋白编码基因,重复序列占比达 62.62%。演化分析表明,显齿蛇葡萄共经历两次全基因组复制(WGD)事件:一次是核心被子植物共有的全基因组三倍化事件,另一次则是葡萄科植物共有的全基因组复制事件。通过转录组 - 代谢组联合分析,本研究鉴定到 AgF3H1 基因 ** 在显齿蛇葡萄二氢杨梅素(一种二氢黄酮醇类物质)的生物合成过程中发挥关键作用。分子对接实验证实,在显齿蛇葡萄的黄酮类化合物合成通路中,AgF3H 基因是催化五羟基黄酮转化为二氢杨梅素的必需基因。据此推测,AgF3H1 是调控显齿蛇葡萄二氢杨梅素合成途径的核心基因 。本研究成果为显齿蛇葡萄的分子育种提供了宝贵的遗传资源,同时也深化了学界对葡萄科植物基因组演化规律,以及药食同源植物黄酮类化合物合成调控机制的理解。
1 引言
显齿蛇葡萄是中国特有的葡萄科植物,作为一种古老的药食同源植物,其天然分布区主要集中于长江以南的广东、湖南、湖北等省份(Gu 等,2020; Cao 等,2023)。《全国中草药汇编》记载,显齿蛇葡萄全株可入药,具清热解毒、平肝降压之功效,临床可用于治疗感冒、发热及肝炎等病症(Ma 等,2019; Li 等,2022)。以显齿蛇葡萄嫩茎叶为原料,经杀青、揉捻、干燥等工序加工制成的 "藤茶",又名 "茅岩莓茶",有止咳化痰、祛风除湿的作用(Carneiro 等,2021; Luo 等,2023)。藤茶的相关记载最早见于《茶经》,起初仅为壮族、瑶族等少数民族的传统饮品,后逐渐被土家族、侗族、客家等族群接纳并推广(Wu 等,2023)。
黄酮类化合物是广泛存在于植物体内的次生代谢产物,不仅在植物的防御反应与生长发育过程中扮演重要角色,还具有极高的保健与药用价值,因此一直是学界的研究热点(Shen 等,2022; Liu 等,2023)。现代药理研究表明,显齿蛇葡萄嫩茎、叶及梢的主要活性成分为黄酮类化合物(Zeng 等,2023)。其总黄酮含量高达 35%~45%,是目前已知黄酮含量最高的植物,具备巨大的商业开发潜力与市场前景(Zhang 等,2016; Zhang X. 等,2019)。二氢杨梅素是一种天然的二氢黄酮醇类化合物,其主要天然来源即为显齿蛇葡萄(Zeng 等,2023)。在显齿蛇葡萄提取物中,二氢杨梅素占总黄酮含量的比例约为 35%,是该植物中含量最丰富的黄酮单体(Zhang 等,2018; Hu 等,2020)。二氢杨梅素具有显著的药理活性,具体表现为降血糖(Chen 等,2016; Ran 等,2019)、抗氧化(Ye 等,2015; Xie 等,2019)、抗肿瘤(Zhou 等,2014; Guo 等,2019)、抗炎(Hou 等,2015; Zhang 等,2022)及抗菌等作用(Wu 等,2017; Xiong 等,2021)。因此,显齿蛇葡萄常被加工为茶叶、饮料、含片等膳食补充剂(Carneiro 等,2020; Zhang 等,2021);此外,显齿蛇葡萄提取物可抑制黑色素生成,被广泛应用于美白护肤产品中(Huang 等,2016)。由此可见,提高显齿蛇葡萄植株的黄酮含量,对提升其药用与营养价值至关重要。
植物黄酮类化合物的合成途径主要包括花青素、异黄酮、黄酮及黄酮醇支路,整个通路由查尔酮合成酶、查尔酮异构酶与黄烷酮 3 - 羟化酶(F3H)共同启动(Zeng 等,2013; Ni 等,2020)。其中,F3H 基因 编码黄酮醇合成途径的关键酶,可催化黄烷酮类物质转化为二氢山奈酚、二氢槲皮素与二氢杨梅素(Prescott & John, 1996)。迄今为止,科研人员已从苹果(Davies, 1993)、葡萄(Sparvoli 等,1994)、拟南芥(Pelletier & Shirley, 1996)及大豆(Zabala & Vodkin, 2005)等多种植物中克隆得到 F3H 基因。目前关于显齿蛇葡萄的研究主要集中于药理活性、抗氧化能力、生理生化特性、转录组测序及叶绿体基因组分析等方面(Ye 等,2015; Huang 等,2016; Gu 等,2020; Luo 等,2023; Wu 等,2023)。基于转录组测序技术,Li 等(2020)与 Yu 等(2021)分别预测了参与显齿蛇葡萄黄酮类化合物及二氢杨梅素合成途径的关键基因;Zhang 等(2022)通过转录组测序鉴定得到一个 AgF3H 基因,从叶片 cDNA 中克隆获得该基因的全长编码序列,并在酿酒酵母中验证了其表达情况,为解析显齿蛇葡萄二氢杨梅素的羟基化修饰机制提供了依据。尽管目前已在显齿蛇葡萄中鉴定出多个参与黄酮合成的相关基因,但黄酮类化合物在该植物不同组织中的积累模式及其分子调控机制仍有待阐明。
本研究以采自湖南省永顺县的野生显齿蛇葡萄(编号 PZY009,图 1a)为实验材料,整合 Illumina 短读长、PacBio 与 ONT 超长读长测序技术,并结合 Hi-C 染色体构象捕获技术,首次完成了显齿蛇葡萄近乎完整的参考基因组组装。同时,选取同一发育阶段的 PZY009 植株根、嫩梢、茎、叶组织(图 1b)开展转录组与代谢组联合分析,旨在鉴定调控黄酮类化合物合成的关键酶基因。本研究结果可为解析显齿蛇葡萄的演化历程、推动分子辅助育种及挖掘活性成分的化学多样性提供重要参考。
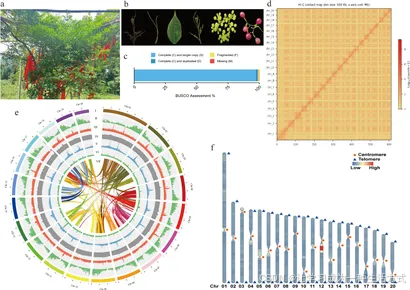
图 1 显齿蛇葡萄基因组精细组装图谱
(a) 显齿蛇葡萄植株形态特征:图中红色标识为当地居民用于祈福的红绸带。(b) 显齿蛇葡萄不同组织器官形态:依次为根、嫩梢、茎、叶、花、果实。(c) 基于 BUSCO 的基因组完整性评估结果。(d) 基因组 Hi-C 互作热图:横坐标为染色体长度,纵坐标为染色体编号,图例代表染色体内互作频率;热图分析窗口大小(bin)为 500 kb。(e) 染色体特征分布图(分辨率为 500 kb):(I) 染色体长度;(II) 基因密度;(III) 转座子(TE)密度;(IV) GC 含量;(V) 长末端重复序列(LTR)密度;(VI) 重复序列覆盖率;(VII) 基因组共线性区块。(f) 端粒与着丝粒鉴定图谱:三角形代表端粒,圆形代表着丝粒;暖色调区域表示转座子(TE)高密度区,冷色调区域表示转座子低密度区。
2 材料与方法
2.1 实验材料与基因组测序
实验材料为采自湖南省湘西土家族苗族自治州永顺县显齿蛇葡萄种植基地的野生种质资源(编号 PZY009),采集其根、茎、叶及嫩梢组织(图 1b),地理坐标为北纬 29°16′42″、东经 109°53′17″。采集后的样品经液氮速冻处理,置于 - 80℃冰箱保存备用。
采用改良 CTAB 法(Allen 等,2006)从显齿蛇葡萄叶片中提取高质量基因组 DNA,通过琼脂糖凝胶电泳与分光光度计检测 DNA 的完整性与纯度。参照 Illumina 标准建库流程构建双端测序文库(2×150 bp),并在 Illumina NovaSeq 测序平台完成测序;利用 PromethION 测序仪开展 ONT 超长读长测序;PacBio 测序则以片段长度≤15 kb 的基因组 DNA 为模板构建长读长文库,基于 PacBio Sequel Ⅱ 平台测序以获得高保真(HiFi)循环一致性测序(CCS)读长;采用 HindⅢ 限制性内切酶构建 Hi-C 测序文库,并在 Illumina NovaSeq 平台完成测序,用于染色体挂载组装。
为开展全转录组分析,采用植物 RNA 提取试剂盒,参照标准流程分别从根、嫩梢、茎、叶组织中提取总 RNA。按照牛津纳米孔技术公司(Oxford Nanopore Technologies)的链交换建库方案富集 mRNA 并合成 cDNA,经 PCR 扩增后利用 PromethION 测序仪完成转录组测序。
2.2 基因组组装与质量评估
基于 Illumina 短读长测序数据,利用 17-mer 频率分布法评估显齿蛇葡萄的基因组大小、杂合度及重复序列含量。对原始 PacBio subreads 进行过滤后,借助 PacBio CCS 分析流程(https://github.com/PacificBiosciences/ccs)完成序列校正,并使用 hifiasm 软件(v0.16.1-r375)(Cheng 等,2021)进行从头组装;利用 Pilon 软件对初步组装得到的 contig 序列进行碱基校正。分别采用 BWA-MEM(v0.7.17)(Li 和 Durbin, 2009)、CEGMA(Parra 等,2007)与 BUSCO(v5.2.2)(Simão 等,2015)评估基因组组装质量。采用基于 DNase 酶切的 Hi-C 建库方法(Ramani 等,2020),在 Illumina NovaSeq 平台进行双端 150 bp 测序以获取染色体构象信息。
处理 Illumina DNA 测序数据时,首先使用 fastp 软件(v0.21.0)过滤低质量读长与接头序列(Chen 等,2018);随后利用基于 k-mer 的分析方法,通过 Jellyfish 软件(v2.2.7)(Marçais 和 Kingsford, 2011)与基因组特征评估软件(GCE)(Liu 等,2013)估算基因组大小与杂合度;为检测测序数据是否存在污染,提取前 50000 条读长,通过 blast + 软件(Camacho 等,2009)与 NT 核苷酸数据库进行比对,并利用 MEGAN 软件(Huson 等,2016)完成物种分类注释。
使用 Filtlong 软件(v0.2.1)与 Porechop 软件(v0.2.4)过滤 ONT 测序数据中的短读长(<10 kb)与接头序列,再利用 NextDenovo 软件对 ONT 超长读长进行初步组装;分别基于 ONT 超长读长与 Racon 软件、Illumina 短读长与 Pilon 软件(v1.24)对 ONT 组装草图进行校正;利用 hifiasm 软件(v0.16.1-r375)结合 PacBio HiFi 读长完成基因组精细组装。
采用 Purge_dups 软件(Guan 等,2020)去除组装序列中的冗余单倍型序列;利用 minimap2 软件(v2.28)(Li, 2018)将组装序列与线粒体、叶绿体参考序列比对,过滤比对率超过 50% 的细胞器序列;通过与 BLAST refseq 数据库比对去除细菌污染序列,同时剔除置信度较低的 contig 序列(McGinnis 和 Madden, 2004)。处理 Hi-C 原始测序数据时,使用 fastp 软件(Chen 等,2018)过滤低质量数据以获得高质量 clean reads;利用 HICUP 软件(Wingett 等,2015)将 clean reads 比对至基因组组装序列,过滤未比对上的读长、无效配对读长及重复读长。
在基因组草图构建过程中,使用 ALLHiC 软件(v0.9.8)(Zhang X. T. 等,2019)通过层次聚类方法构建染色体水平基因组组装图谱;利用 3D-DNA 软件(Dudchenko 等,2017)与 Juicer 软件(v1.5)(Durand 等,2016b)将 contig 间的互作关系转换为特定二进制文件,并通过 Juicebox 软件(Durand 等,2016a)可视化互作热图,指导 contig 序列的人工排序与定向;基于互作关系人工剔除冗余 contig 序列,使用 100 个连续 N 碱基填充序列间隙;利用 HiCExplorer 软件(Wolff 等,2020)绘制 contig 序列间的互作强度与位置关系图谱。
2.3 端粒与着丝粒鉴定
为完成参考基因组的补洞与精细组装,使用 winnowmap 软件(v1.11)(Jain 等,2020),设置参数 k=15 与--MD,将补洞测序数据比对至基因组间隙区域以填补序列缺口;若比对序列覆盖间隙两端,则选取最长且比对质量最高的序列替换间隙区域。随后使用 Winnowmap2 软件(参数设置:k=15、序列相似度 > 0.9998、-MD、ax map-pb)(Jain 等,2022),将填补缺口后的基因组序列与长度≥10 kb 的 PacBio HiFi 读长进行比对。搜索所有读长末端的端粒重复序列(5′端与 3′端均为 AAACCCT),选取重复序列含量最高的读长作为参考序列,其余读长作为查询序列;利用 medaka_consensus 软件对参考序列与查询序列进行组装;通过 nucmer 软件(v3.1)(Kurtz 等,2004)替换各伪染色体末端序列;最后利用 Racon 软件结合 PacBio HiFi 读长对近乎完整的参考基因组进行误差校正。分别使用 CentIER 软件(v3.0)(Xu 等,2024)与端粒鉴定工具包(tidk, v0.2.63)(Brown 等,2025),采用默认参数完成着丝粒与端粒的鉴定。
2.4 基因组注释
分别利用全基因组微卫星分析工具(GMATA, v21)与串联重复序列查找工具(TRF, v4.10)(Benson, 1999)完成串联重复序列注释。整合从头预测 与同源比对 两种方法,对显齿蛇葡萄基因组中的转座子(TE)进行注释:使用 MITE-hunter 软件(Han 和 Wessler, 2010)与 RepeatModeler2 软件(v1.0.11)(Flynn 等,2020),采用默认参数构建显齿蛇葡萄基因组的从头重复序列文库;利用 LTRharvest 软件(Ellinghaus 等,2008)与 LTR_Finder 软件构建长末端重复序列逆转录转座子(LTR-RT)文库,并通过 LTR_retriever 软件(Ou 和 Jiang, 2018)去冗余以获得非冗余 LTR-RT 文库;将上述文库与 TEclass repbase 数据库(v20170127)(Zhuo 和 Feschotte, 2015)比对,完成重复序列家族分类;合并 LTR_retriever、MITE-Hunter 与 RepeatModeler2 构建的重复序列文库,输入至 RepeatMasker 软件(v4.0.7)(Chen, 2004)完成基因组重复序列注释。采用 LTR_retriever 软件,默认参数估算 LTR、Copia 与 Gypsy 类转座子的插入时间。
整合同源比对 、从头预测 与转录组辅助注释 三种方法进行基因结构注释:同源预测方面,利用 blast + 软件(Camacho 等,2009)将近缘物种蛋白序列比对至参考基因组,再通过 Exonerate 软件预测转录本与编码区序列(Slater 和 Birney, 2005);同时将基因组质量评估阶段通过 BUSCO 软件预测的基因整合至同源预测结果中(Manni 等,2021)。从头预测方面,基于训练集数据,分别使用 Augustus 软件(v3.3)(Stanke 等,2008)与 GlimmerHMM 软件(Delcher 等,2007),采用默认参数进行基因预测。转录组辅助注释方面,对于 Illumina RNA-seq 数据,利用 fastp 软件(Chen 等,2018)过滤低质量读长后,通过 HISAT2 软件(Kim 等,2019)将 clean reads 比对至参考基因组;比对结果经 Stringtie 软件拼接获得转录本序列(Kovaka 等,2019),再通过 TransDecoder 软件预测编码区序列。对于 Nanopore RNA-seq 数据,利用 NanoFilt 软件(v2.8.0)与 Pychopper 软件过滤低质量读长并识别全长转录本序列;经 Racon 软件校正后,通过 minimap 软件将全长转录本比对至参考基因组(Li, 2016);比对结果经 Stringtie 软件拼接获得转录本序列(Kovaka 等,2019)。
利用 MAKER 软件(Holt 和 Yandell, 2011)整合上述三种方法预测的基因集,并进行优化以获得最终的蛋白编码基因集;通过 BUSCO 软件(Simão 等,2015)验证基因组注释的完整性,以确保注释结果的可靠性与准确性。
采用 DIAMOND 软件(Buchfink 等,2015)将预测的蛋白序列与多个公共数据库比对以完成功能注释,涉及数据库包括非冗余蛋白数据库(NR)(Deng 等,2006)、瑞士蛋白数据库(Swiss-Prot)(Boeckmann 等,2003)、直系同源基因数据库(eggNOG)、基因本体数据库(GO)及京都基因与基因组百科全书(KEGG)(Kanehisa 和 Goto, 2000);通过上述比对分析,挖掘基因的潜在功能、保守基序与蛋白结构域。利用 KOBAS 软件完成 KEGG 通路注释(Xie 等,2011);采用 InterProScan 软件,默认参数预测基因的结构域与 GO 功能条目(Blum 等,2021)。利用 BLAST + 软件(Camacho 等,2009)将经 EvidenceModeler 软件整合的蛋白序列(Haas 等,2008)与四大公共蛋白数据库比对,设置 E 值阈值为 1e-05,保留 E 值最低的比对结果。
非编码 RNA(ncRNA)分为微小 RNA(miRNA)、核糖体 RNA(rRNA)、转运 RNA(tRNA)、核仁小 RNA(snoRNA)与核内小 RNA(snRNA)等类别。采用数据库比对 与模型预测 两种策略进行非编码 RNA 注释:使用 tRNAscan-SE 软件,设置真核生物参数,预测 tRNA 序列(Chan 等,2021);利用 Infernal cmscan 软件(Nawrocki 和 Eddy, 2013),基于 Rfam 数据库比对鉴定 miRNA、rRNA、snRNA 与 snoRNA 序列;通过 RNAmmer 软件(Lagesen 等,2007)预测 rRNA 及其亚基序列。
2.5 比较基因组学分析
为保证比较基因组学分析的准确性,选取基因组组装质量高且与显齿蛇葡萄系统发育关系近缘的物种作为研究对象,包括葡萄科的 3 个葡萄属物种(欧洲葡萄、圆叶葡萄、山葡萄)与白粉藤属物种(圆叶白粉藤);同时纳入模式植物(拟南芥、水稻)及多种高黄酮含量植物(大豆、罂粟、丹参、番茄、铁皮石斛、黄芩);基于系统发育距离,选取单子叶植物(蓝星睡莲)作为外类群(补充表 S13)。利用 blast + 软件(Camacho 等,2009)与 OrthoFinder 软件(Emms 和 Kelly, 2019)对上述 14 个物种进行基因家族聚类分析;通过 Panther 数据库完成基因家族功能注释(Mi 等,2019);利用 clusterProfiler 软件(Yu 等,2012)对各物种特有基因家族进行 GO 与 KEGG 富集分析。提取单拷贝直系同源基因,通过 MUSCLE 软件(Edgar, 2004)进行多序列比对;利用 TrimAl 软件(Capella-Gutiérrez 等,2009)过滤比对结果,构建超级矩阵用于后续分析。
基于超级矩阵,使用 RAxML 软件,采用 PROTGAMMAWAG 模型构建最大似然(ML)系统发育树(Stamatakis, 2014);利用 PAML 软件中的 MCMCTree 程序(Yang, 2007)估算物种分化时间,参数设置为:burn-in=10000、抽样次数 = 100000、抽样频率 = 2,分化时间校准点取自 TimeTree 数据库(Kumar 等,2022),具体校准时间如下:蓝星睡莲 - 水稻:1.68-1.91 亿年前;铁皮石斛 - 水稻:1.08-1.23 亿年前;罂粟 - 水稻:1.42-1.63 亿年前;罂粟 - 拟南芥:1.26-1.36 亿年前;欧洲葡萄 - 拟南芥:1.09-1.24 亿年前;大豆 - 拟南芥:1.02-1.12 亿年前;番茄 - 拟南芥:1.11-1.23 亿年前;番茄 - 丹参:0.75-0.96 亿年前;黄芩 - 丹参:0.33-0.72 亿年前;欧洲葡萄 - 圆叶白粉藤:0.31-0.96 亿年前;欧洲葡萄 - 圆叶葡萄:0.04-0.14 亿年前;欧洲葡萄 - 山葡萄:0.05-0.40 亿年前。
使用 DIAMOND 软件(Buchfink 等,2015)对 14 个物种的蛋白组进行全基因组比对,设置 E 值阈值为 10-5;利用 PhyloMCL 软件(Zhou 等,2020),默认参数获取同源基因家族(HOG);对每个同源基因家族,通过 PASTA 软件进行多序列比对(Tang 和 Riva, 2013),并将蛋白比对结果转换为核苷酸比对结果;利用 IQ-TREE2 软件(Minh 等,2020),设置 100 次自举检验,为每个同源基因家族构建最大似然树;对于自举值 > 50% 的节点,采用已报道的分析策略估算基因树的基因重复与丢失事件(Ren 等,2018);统计基因重复候选事件的重复基因保留模式,用于后续分析。
将基因家族演化模型设定为随机生死过程,演化速率为每百万年 1 个基因的扩张或收缩速率;利用 CAFE 软件(Han 等,2013)预测显齿蛇葡萄相对于其祖先的基因家族变化,设置 P 值阈值为 0.05 以筛选显著扩张或收缩的基因家族;结合系统发育树拓扑结构与分支长度,分析基因家族变化的显著性。
对单拷贝直系同源基因进行 MUSCLE 多序列比对(Edgar, 2004);利用 PAML 软件中的 CodeML 程序(Yang, 2007),将显齿蛇葡萄设为前景分支,进行正选择分析;采用 χ² 检验计算 P 值,并通过错误发现率(FDR)校正以控制多重检验误差。
共线性分析流程如下:利用 DIAMOND 软件(Buchfink 等,2015)鉴定物种间的同源基因对,设置参数 E 值 <1e-5、共线性评分(C-score)>0.5,并通过 JCVI 软件过滤(Tang 等,2024);基于 gff3 文件确定染色体上相邻的同源基因对;利用 MCScanX 软件,设置参数 - a -e 1e-5 -s 5,识别共线性区块(Wang 等,2012);通过 R 语言 circlize 包绘制基因组共线性环形图(Gu 等,2014);使用命令jcvi.compara.catalog orthologs --cscore=0.7识别共线性区块(Tang 等,2024),并提取所有共线性区块中的基因序列。
为鉴定显齿蛇葡萄基因组中的全基因组复制(WGD)事件,整合全基因组复制检测工具、基因组共线性分析、同义替换率(Ks)估算与峰值拟合等方法(Sun 等,2022);联合分析共线性区域的 4 倍简并位点转换率(4DTv)与同义替换率(Ks),是目前鉴定全基因组复制事件的通用方法;利用 WGD 软件(Zwaenepoel 和 Van de Peer, 2019)完成显齿蛇葡萄全基因组复制事件的鉴定。
2.6 转录组数据分析
对原始 RNA 短读长数据进行预处理:过滤接头序列、去除 poly (A) 尾序列、剔除低质量读长(Q<20);统计高质量 clean reads 的 Q20、Q30 值与 GC 含量;利用 HISAT 软件将 clean reads 比对至参考基因组与全长转录本序列(Kim 等,2019);选取完全匹配或存在单个碱基错配的比对读长,通过 StringTie 软件重构转录本序列(Kovaka 等,2019)。基于比对结果鉴定表达基因:若读长比对至已注释基因序列,则判定该基因为已知基因;若读长仅比对至全长转录本序列,未匹配任何已注释基因,则判定为新基因并进行注释。
采用每千碱基转录本每百万映射读长(FPKM)值量化基因表达水平;利用 edgeR 软件,通过缩放因子对测序文库的读长计数进行标准化(Robinson 等,2010);使用 EBSeq 软件分析同源基因在根、茎、叶、嫩梢 4 种组织中的差异表达模式;利用 DESeq2 软件(Anders 和 Huber, 2010)进行组织间的差异表达分析;设置筛选条件为错误发现率(FDR)<0.05 且 | log2 (倍数变化)|≥2,鉴定显著差异表达基因(DEG);利用 clusterProfiler 软件对差异表达基因进行 GO 与 KEGG 富集分析(Wu 等,2021);通过 NetworkAnalyst 软件与 STRING 数据库构建差异基因的蛋白质 - 蛋白质相互作用(PPI)网络。
2.7 广泛靶向代谢组分析
利用 ProteoWizard 软件包中的 MSConvert 工具,将原始代谢组数据转换为 mzXML 格式(Rasmussen 等,2022);在 R 语言环境中通过 XCMS 软件完成代谢物特征检测、保留时间校正与峰对齐(Navarro-Reig 等,2015)。通过精确质量数与二级质谱(MS/MS)数据,结合多个数据库进行代谢物鉴定,涉及数据库包括人类代谢组数据库(HMDB)(Wishart 等,2007)、质谱数据库(MassBank)(Horai 等,2010)、Knapsack 数据库、ReSpect 数据库、脂质图谱数据库(LipidMaps)(Sud 等,2007)、KEGG 数据库及中国苏州帕诺米克生物医药科技有限公司的专有数据库。基于母离子的质荷比(m/z)确定代谢物分子量;结合百万分比误差(ppm)与加合离子信息预测代谢物分子式,并与数据库比对完成一级质谱鉴定;同时匹配二级质谱碎片离子与数据库信息,完成代谢物的二级质谱鉴定。
采用两种多元统计分析模型区分样本组间差异,包括无监督主成分分析(PCA)与有监督偏最小二乘判别分析(PLS-DA、OPLS-DA),分析过程通过 R 语言 ropls 包完成(Thévenot 等,2015);基于组间比较的 P 值判断差异显著性;结合 P 值、正交偏最小二乘判别分析的变量投影重要性(VIP)与倍数变化筛选差异代谢物;设置筛选条件为 P<0.05 且 VIP>1,鉴定显著差异积累代谢物。
利用 MetaboAnalyst 软件(Xia 和 Wishart, 2011)对差异代谢物进行通路富集分析,整合通路富集分析与拓扑结构分析两种方法;将鉴定的差异代谢物映射至 KEGG 通路,进行生物学功能解析;通过 KEGG Mapper 工具绘制代谢通路可视化图谱。
2.8 加权基因共表达网络分析
为鉴定高相关性的基因共表达模块,利用 R 语言 WGCNA 包进行加权基因共表达网络分析(WGCNA)(Zhang 和 Horvath, 2005);将邻接矩阵转换为拓扑重叠矩阵(TOM),通过 WGCNA 软件的 goodGenes 函数过滤低质量基因;利用 cutreeDynamic 函数修剪基因层次聚类树,并合并相关系数(r)>0.75 的共表达模块;采用无符号拓扑重叠矩阵(unsigned TOMType),通过 blockwiseModules 函数构建基因共表达网络。利用 WGCNA 软件的 module eigengenes 函数计算模块特征基因,并通过皮尔逊相关分析评估模块特征基因与表型性状的关联性。利用 Cytoscape 软件中的 CytoHubba 插件(Chin 等,2014)鉴定共表达网络中的核心基因(Hub gene)(Shannon 等,2003)。
2.9 AgF3H 基因的分子对接分析
利用 AlphaFold3 软件预测显齿蛇葡萄 AgF3H 蛋白的晶体结构(Abramson 等,2024);通过 Schrödinger 软件中的 Protein Preparation Wizard 模块对晶体结构进行预处理,包括天然配体状态还原、氢键优化、能量最小化与水分子去除(Abramson 等,2024)。利用 Schrödinger 软件中的 LigPrep 模块,将五羟基黄烷酮、柚皮素与圣草酚的二维 sdf 结构文件转换为三维手性构象。通过 SiteMap 模块确定蛋白的最优结合位点;利用 Receptor Grid Generation 模块为该结合位点构建最优包围盒,从而定义 AgF3H 蛋白的活性位点。采用高精度 XP 对接方法,将五羟基黄烷酮、柚皮素与圣草酚分别对接至 AgF3H1 与 AgF3H2 蛋白的活性位点。通过分子力学 - 广义波恩表面积模型(MM-GBSA)计算配体与蛋白的结合自由能(dG Bind),结合自由能数值越低,表明配体与蛋白的结合越稳定。
参照试剂盒说明书,使用 M-MLV 反转录酶试剂盒合成第一链 cDNA,用于实时荧光定量 PCR(qRT-PCR)分析。qRT-PCR 反应体系为 iTaq Universal SYBR Green 超级混合液,通过 ABI 7500 PCR 系统完成扩增与信号检测。实验设置 3 次生物学重复,每个重复均包含标准品与阴性对照。qRT-PCR 反应程序如下:95℃预变性 30 s;95℃变性 5 s、60℃退火 30 s、60℃延伸 20 s,共 40 个循环。每个样品设置 3 次技术重复,qRT-PCR 结果取 3 次重复的平均值。以显齿蛇葡萄甘油醛 - 3 - 磷酸脱氢酶基因(GAPDH)作为内参基因(Xu, 2017),通过 Ct 值计算 AgF3H1 基因的相对表达量(Livak 和 Schmittgen, 2001)(补充表 S21)。
3 结果
3.1 显齿蛇葡萄基因组测序与组装
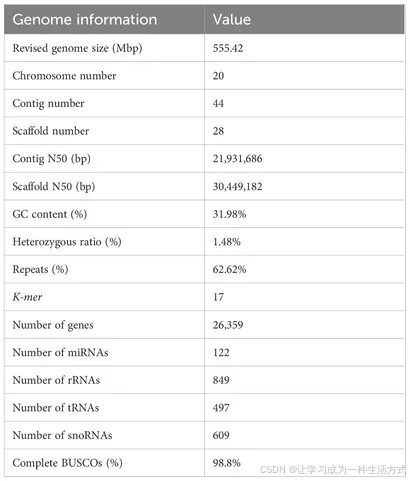
为估算显齿蛇葡萄的基因组大小,本研究基于 Illumina 测序平台,获得了 70.8 Gb 的高质量双端测序数据,并利用 17-mer 分析方法进行评估(表 1;补充图 S1;补充表 S1)。最终组装得到的显齿蛇葡萄基因组大小为 555.42 Mb,GC 含量为 31.98%,重复序列占比 62.62%,杂合度为 1.48%(表 1、补充表 S1),这表明该物种基因组具有高杂合、高重复 的特征。
本研究通过序列一致性分析与 BUSCO 评估两种方法,对基因组组装质量进行验证。序列一致性分析结果显示,短读长序列与显齿蛇葡萄基因组的比对率达 99.02%,覆盖率达 99.29%,说明组装序列的一致性较高(补充表 S2)。BUSCO 评估基于 425 个单拷贝直系同源基因集开展,结果显示基因组完整性为 98.8%,证实本次组装的基因组具有较高的完整性(图 1c)。
显齿蛇葡萄基因组中碱基 A、T、G、C 的占比均处于正常范围,N 碱基含量为 0.00%,远低于可接受阈值(<10%)(补充表 S3)。基因组杂合单核苷酸多态性(SNP)比例为 0.2953%,纯合 SNP 比例仅为 6.7836e-05%(补充表 S4),表明组装序列的单碱基准确性较高。上述结果证实,本研究组装的显齿蛇葡萄基因组序列兼具高一致性、高准确性与高完整性。
3.2 Hi-C 技术辅助组装显齿蛇葡萄近乎完整的参考基因组
为实现染色体水平的基因组组装,本研究利用高通量染色体构象捕获(Hi-C)测序技术,共获得 69.8 Gb 的测序数据,对应 2.3536 亿条双端 Hi-C 读长(补充表 S1)。借助 ALLHiC 软件,将总计 608.41 Mb 的 28 个 scaffold 序列挂载至显齿蛇葡萄基因组上(补充表 S5)。通过 Hi-C 技术的辅助纠错与组装优化,最终获得 20 条染色体水平的序列,基因组挂载率高达 99.89%(补充表 S6)。每条染色体至少包含 1 个 scaffold,长度范围介于 18.57 Mb(Chr 20)至 59.11 Mb(Chr 1)之间(补充表 S7)。
Hi-C 互作矩阵热图结果显示,染色体内相邻序列间的互作强度更高,20 条伪染色体沿对角线呈规则分布(图 1d)。基于葡萄基因组数据,绘制了显齿蛇葡萄基因组的共线性环形图(图 1e)。以 7 碱基端粒重复序列(AAACCCT)为探针,在 20 条伪染色体上共鉴定出 38 个端粒区域(仅 Chr 03 与 Chr 17 各缺失 1 个端粒),并在每条染色体上定位了潜在的着丝粒区域,具体位置信息详见补充表 S8 与 S9。综合以上指标,本次组装的基因组达到了高质量、近乎完整的标准(图 1f)。
3.3 重复序列预测与基因组注释
真核生物基因组中的重复序列在物种演化、遗传变异及生命活动调控中发挥关键作用,对全面解析基因表达调控机制、基因组结构特征与物种演化规律具有重要意义。本研究整合同源比对、从头预测与转录组辅助注释 三种方法,在显齿蛇葡萄基因组中共预测得到 25756 个基因,基因数量多于蓝星睡莲(19299 个),但少于欧洲葡萄(29591 个)、圆叶葡萄(26742 个)与山葡萄(29168 个)(补充图 S2a)。
显齿蛇葡萄基因组的平均基因长度为 7895 bp,平均外显子长度为 351 bp,平均内含子长度为 1241 bp,平均编码区长度为 1615 bp(补充表 S10)。同源基因功能注释结果显示,分别有 9848 个基因(37.36%)与 25990 个基因(98.60%)在 eggNOG 与 NR 数据库中获得同源匹配(补充表 S11;补充图 S2b)。此外,本研究在显齿蛇葡萄基因组中鉴定出 2077 个非编码 RNA,包括 849 个核糖体 RNA(rRNA)、497 个转运 RNA(tRNA)、122 个微小 RNA(miRNA)与 609 个核仁小 RNA(snoRNA)(补充表 S12)。
3.4 比较基因组学分析
本研究选取 12 个近缘物种(欧洲葡萄、圆叶葡萄、山葡萄、圆叶白粉藤、拟南芥、大豆、铁皮石斛、罂粟、丹参、番茄、水稻、黄芩)与 1 个外类群物种(蓝星睡莲),将其基因组与显齿蛇葡萄基因组进行比较分析(补充表 S13)。基于这些物种的基因组数据,开展同源基因鉴定、基因家族聚类,并分析显齿蛇葡萄中富集的单拷贝基因(图 2a)。
分析结果显示,14 个物种共包含 27473 个基因家族,其中保守基因家族数量为 6738 个,涵盖所有物种共享的 175 个单拷贝基因家族(图 2a)。选取圆叶白粉藤、拟南芥、欧洲葡萄、圆叶葡萄、山葡萄、显齿蛇葡萄与番茄的基因家族聚类数据,绘制了基因家族交集的 upset 图(图 2b)。该图显示,相较于其他物种,显齿蛇葡萄拥有 193 个特有基因家族,共包含 1075 个基因。富集分析结果表明,这些特有基因家族主要参与代谢途径 与转运过程 (补充图 S3)。
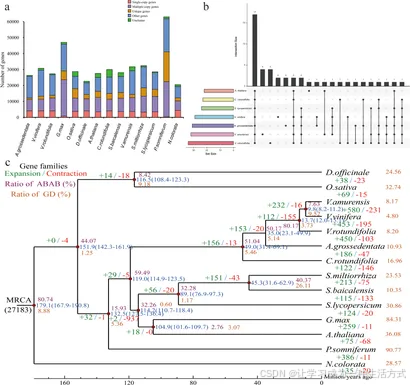
图 2 基因家族聚类与演化分析
(a) 14 个物种的基因家族拷贝数分布:涉及物种包括欧洲葡萄、圆叶葡萄、山葡萄、圆叶白粉藤、拟南芥、大豆、铁皮石斛、罂粟、丹参、番茄、水稻、黄芩、蓝星睡莲及显齿蛇葡萄。(b) 显齿蛇葡萄与欧洲葡萄、山葡萄、圆叶葡萄、拟南芥、圆叶白粉藤、番茄的基因家族共有及特有情况的 Upset 图。(c) 14 个物种的系统发育树、分化时间及基因家族扩张 / 收缩分析:通过基因树与物种树的联合解析,鉴定不同真双子叶植物分支的基因重复(GD)事件;节点数值代表估算的分化时间(单位:百万年前,Mya),蓝色括号内为误差范围;红色数值与绿色数值分别表示基因家族收缩数量与扩张数量。
基于 14 个物种共有的 175 个单拷贝同源基因,本研究采用贝叶斯松弛分子钟法 构建了高置信度的系统发育树,并估算了物种间的分化时间(图 2c)。在这 14 个物种中,显齿蛇葡萄与圆叶白粉藤的亲缘关系最近,二者的分化时间约为 49.0 百万年前;显齿蛇葡萄支系与葡萄属(欧洲葡萄、圆叶葡萄、山葡萄)支系则在约 35.0 百万年前从共同祖先分化而来(图 2c)。
基因家族的扩张与收缩是植物形成物种特异性性状、产生表型多样性的关键驱动力,其中扩张的基因家族可能会获得新功能,进而提升植物的环境适应性。分析结果显示,这 14 个物种的最近共同祖先(MRCA)共包含 27183 个基因家族(图 2c)。与近缘祖先相比,显齿蛇葡萄基因组中有186 个基因家族发生显著扩张 (包含 1097 个基因),同时有47 个基因家族发生收缩 (包含 127 个基因)(图 2c)。GO 富集分析结果表明,收缩的基因家族主要富集于刺激响应 与甲基化 相关通路(补充图 S4a);而扩张的基因家族则显著富集于次生代谢物生物合成过程 与次生代谢过程 通路(补充图 S4b)。
3.5 显齿蛇葡萄的全基因组复制事件分析
对葡萄科代表性物种(圆叶白粉藤、欧洲葡萄、圆叶葡萄、山葡萄)与显齿蛇葡萄的共线性深度分析显示,二者的共线性深度比值接近 1:1,且共线性模式高度保守。这表明显齿蛇葡萄与上述葡萄科物种的祖先支系均经历了近期与古老的全基因组复制(WGD)事件 (图 3a--d)。
基于 14 个物种的基因组数据开展 Tree2GD 分析,结果显示:葡萄目(Vitales Juss. ex Bercht. & J. Presl)祖先发生过 2 次多倍化事件,对应 11681 个基因重复(GD)事件;蔷薇目(Rosales Bercht. & J. Presl)祖先同样发生多倍化事件,对应 15800 个基因重复事件。进一步分析发现,葡萄目祖先的(AB)(AB)型基因重复比例为 50.17%,蔷薇目祖先的该比例为 51.14%,这为全基因组复制事件的发生提供了强有力的系统发育基因组学证据。
对 14 个物种的基因组进行 Tree2GD 分析发现,显齿蛇葡萄基因组中总共鉴定出 20029 个基因重复事件,占比 10.93%,这一结果证实显齿蛇葡萄演化历程中经历了两次多倍化事件 (图 2c)。
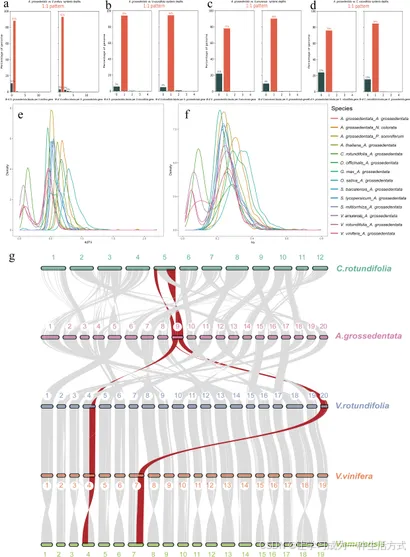
图 3 显齿蛇葡萄比较基因组与演化分析
(a--d) 葡萄科代表性物种(圆叶白粉藤、欧洲葡萄、圆叶葡萄、山葡萄)与显齿蛇葡萄的共线性深度分析。(e, f) 14 个物种的 4 倍简并位点颠换率(4DTv)与同义替换率(Ks)分布:虚线代表显齿蛇葡萄 4DTv 与 Ks 分布的峰值;4DTv,4 倍简并密码子第三位颠换率;Ks,同义替换率。(g) 圆叶白粉藤、圆叶葡萄、欧洲葡萄、山葡萄与显齿蛇葡萄的基因组共线性分析:灰色线条代表物种间的共线性区块;红色线条代表显齿蛇葡萄与上述 4 个葡萄科物种之间的染色体片段易位事件。
4DTv 与 Ks 的分布图谱呈现出两个明显的峰值 ,这一结果表明显齿蛇葡萄演化历程中发生过两次全基因组复制(WGD)事件(图 3e, f)。其中较近期的一次全基因组复制事件,与圆叶白粉藤、欧洲葡萄、圆叶葡萄、山葡萄的全基因组复制事件相吻合,证实这是葡萄科物种共有的一次全基因组复制事件;另一次全基因组复制事件则对应核心真双子叶植物共有的全基因组三倍化事件(WGT,即 γ 事件)(Jaillon 等,2007)。
为解析葡萄科物种的染色体演化规律与系统发育关系,本研究对圆叶白粉藤、欧洲葡萄、圆叶葡萄、山葡萄及显齿蛇葡萄开展了基因组共线性分析(图 3g)。结果显示,显齿蛇葡萄与圆叶白粉藤的共线性图谱中散点较少,说明二者的系统发育关系相较于其他物种更为紧密(图 3g)。此外,本研究在显齿蛇葡萄的 9 号染色体上检测到重组与基因片段重排事件,包括染色体倒位与易位(图 3g),这些变异可能是导致显齿蛇葡萄黄酮类化合物高含量的重要原因。综上,研究结果为解析显齿蛇葡萄的染色体演化机制提供了新视角,同时也为跨属研究葡萄属植物重要农艺性状提供了科学依据。
3.6 代谢组与转录组联合分析
黄酮类化合物广泛存在于葡萄科植物中(包括显齿蛇葡萄),在植物次生代谢过程中发挥多种生物学功能。前期研究已鉴定出 138 个黄酮合成相关基因及异构体,初步解析了黄酮类化合物的生物合成通路(Li 等,2020)(图 4a)。
本研究对显齿蛇葡萄的根、嫩梢、茎、叶 4 种组织开展代谢组分析,旨在明确黄酮类化合物积累与基因表达的关联性。对 3 次生物学重复样本的主成分分析(PCA)、正交偏最小二乘判别分析(OPLS-DA)及偏最小二乘判别分析(PLS-DA)结果证实,测序数据具有良好的重复性(补充图 S5)。基于高分辨率液相色谱 - 串联质谱(LC-MS/MS)分析,在 4 种组织中总共检测到 1526 种代谢物,其中包括黄酮类化合物 70 种、萜类化合物 41 种、羧酸及其衍生物 37 种(补充图 S6;补充表 S14)。在 70 种黄酮类化合物中,有 35 种属于黄酮生物合成通路(Ko00941)的核心代谢物,占比达 50%(补充表 S15)。除 4 种特异地积累于根中的代谢物(8 - 异戊烯基柚皮素、松属素、柯因 D、芹菜素)外,其余黄酮类化合物主要在嫩梢组织中高表达(图 4b)。
已有研究表明,显齿蛇葡萄的黄酮合成主要依赖二氢杨梅素的积累,涉及的关键代谢物包括柚皮素、二氢杨梅素、杨梅素与飞燕草素等(Yu 等,2021)。嫩梢与根的差异代谢物富集分析结果显示,差异代谢物在黄酮生物合成通路中富集程度最高(补充图 S7);其中二氢杨梅素在嫩梢组织中的含量最高,且在嫩梢与根之间的差异表达水平最为显著(图 4c)。
基于本研究组装的近乎完整的显齿蛇葡萄基因组,对 PZY009 株系的根、茎、叶、嫩梢组织开展转录组测序,旨在鉴定参与二氢杨梅素合成的关键基因与转录因子(TF)。3 次生物学重复样本的 PCA 分析结果证实,测序数据重复性良好(补充图 S8)。基因表达差异分析显示,嫩梢与根组织相比共有 10566 个差异表达基因,其中上调基因 5993 个、下调基因 4573 个(补充表 S16;补充图 S9)。GO 与 KEGG 富集注释结果表明,这些差异基因的功能主要富集于代谢途径与次生代谢物合成通路,其中以黄酮类化合物合成通路与异黄酮合成通路的富集程度最为显著(补充图 S10)。
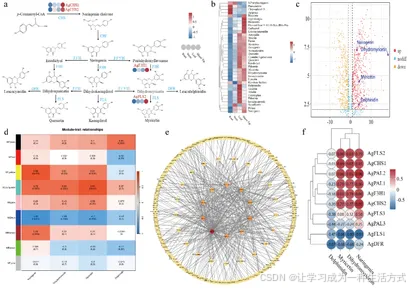
图 4 代谢物含量、基因表达聚类及黄酮类化合物生物合成通路分析
(a) 显齿蛇葡萄黄酮类化合物生物合成通路简化图:蓝色字体标注催化酶;圆角矩形标注基因表达量。(b) 同一发育阶段下,黄酮合成通路中 35 种代谢物在不同组织中的表达量热图:以标准化(z-score)的 FPKM 值为依据,用颜色梯度表示表达水平高低。(c) 根与嫩梢中差异代谢物的火山图:横坐标为差异倍数的对数(Log₂ fold change);纵坐标为显著性 P 值的负对数(-log₁₀ P-value);散点大小代表变量投影重要性(VIP)值;红色代表上调代谢物,黄色代表下调代谢物,蓝色代表无显著差异的代谢物;标注的代谢物包括柚皮素、二氢杨梅素、杨梅素、飞燕草素。(d) 基因表达模块与黄酮类代谢物的相关性热图:行代表不同的共表达模块(ME);列代表黄酮类化合物。(e) 基于 Cytoscape 软件构建的共表达网络:展示棕色、黄色、蓝色、青绿色模块中 10 个核心枢纽基因与 240 个转录因子的互作关系。(f) 加权基因共表达网络分析(WGCNA)筛选出的 10 个关键基因,与柚皮素、二氢杨梅素、杨梅素、飞燕草素的相关性热图。
3.7 黄酮类化合物生物合成通路关键基因筛选
在黄酮合成通路(Ko00941)富集的 35 种代谢物中,本研究鉴定出 4 种核心代谢物:柚皮素、二氢杨梅素、杨梅素与飞燕草素。基于此,本研究结合转录组数据,以这 4 种核心代谢物为目标性状开展加权基因共表达网络分析(WGCNA)。
通过 WGCNA 分析,将 12 份样本中的 17085 个基因聚类为 9 个共表达模块(图 4d;补充图 S11;补充表 S17)。本研究重点关注其中 4 个模块:黄色模块(2707 个基因)、棕色模块(3234 个基因)、青绿色模块(4263 个基因)及蓝色模块(4012 个基因)(图 4d;补充表 S17)。对各模块内基因与转录因子进行相关性分析,以相关系数绝对值≥0.9 为阈值,从黄酮合成通路中筛选得到 10 个关键酶基因与 364 个转录因子,具体分布为:黄色模块(6 个基因、64 个转录因子)、棕色模块(2 个基因、99 个转录因子)、青绿色模块(1 个基因、77 个转录因子)、蓝色模块(1 个基因、124 个转录因子)(补充表 S18、S19)。
显齿蛇葡萄组织特异性表达谱分析结果显示,筛选出的 10 个关键基因具有显著的空间表达异质性。其中,AgF3H1 基因在嫩梢组织中表现出极强的组织特异性表达特征 ,其转录水平(FPKM 值)显著高于其他检测组织(P<0.05),差异具有统计学意义。对次生代谢通路关键结构基因的系统性分析进一步发现,这些基因存在明显的组织表达偏好性:AgFLS1 与AgDFR 基因在根组织中高丰度表达;AgPAL3 与AgFLS3 基因则在茎组织中特异性高表达(补充图 S12)。后续实时荧光定量 PCR(qRT-PCR)验证结果显示,这些基因的组织特异性表达模式与转录组分析数据高度一致(补充图 S13)。
进一步对 10 个关键基因与 364 个转录因子进行组间相关性分析,以相关系数绝对值≥0.95 为阈值,筛选得到 240 个转录因子(补充表 S18)。利用 Cytoscape 软件计算这 10 个基因与 240 个转录因子的连接度(degree)值,选取连接度≥5 的 21 个转录因子进行后续分析(图 4e;补充表 S19)。
显齿蛇葡萄是已知黄酮含量最高的植物,其二氢杨梅素的含量变化趋势基本可以反映总黄酮含量的变化规律。10 个关键基因与柚皮素、二氢杨梅素、杨梅素、飞燕草素的组间相关性热图结果显示,AgF3H1、AgFLS2、AgCHS1、AgCHS2、AgPAL1、AgPAL2 基因与柚皮素、二氢杨梅素、杨梅素的相关性最高 (图 4f)。
在显齿蛇葡萄中,二氢杨梅素的合成由两类酶基因共同催化:一类是AgF3'5'H 基因(催化二氢槲皮素或二氢山奈酚生成目标产物),另一类是AgF3H 基因(催化五羟基黄烷酮生成目标产物)(图 4a)。结合 WGCNA 分析结果,本研究筛选得到 1 个AgF3H 家族基因 ------AgF3H1 ,据此推测AgF3H1 是调控二氢杨梅素合成的关键基因 。
3.8 AgF3H 基因的分子对接分析
黄烷酮 3 - 羟化酶(F3H)属于2 - 酮戊二酸依赖型双加氧酶(2-ODD)家族 ,其蛋白结构具有保守的 N 端区域,C 端结构则与 4 - 羟化酶 α 亚基相似(Cheng 等,2014; Kawai 等,2014)。在显齿蛇葡萄黄酮类化合物生物合成通路中,AgF3H 基因的催化功能具有底物特异性:分别催化圣草酚生成二氢槲皮素、柚皮素生成二氢山奈酚、五羟基黄烷酮生成二氢杨梅素(图 4a)。
通过显齿蛇葡萄基因组序列分析,本研究鉴定出 2 个F3H 家族基因,分别命名为AgF3H1 与AgF3H2 。随后,以圣草酚、柚皮素、五羟基黄烷酮为底物,分别与AgF3H1 和AgF3H2 的氨基酸序列进行分子对接分析。高精度 XP 对接与分子力学 - 广义波恩表面积模型(MM-GBSA)分析结果显示:
- 圣草酚、五羟基黄烷酮与AgF3H1 的对接分数分别为 - 8.032 和 - 6.121,MM-GBSA 结合自由能分别为 - 32.96 kcal/mol 和 - 41.49 kcal/mol,表明二者与AgF3H1 均能稳定结合(补充表 S20)。圣草酚可深入AgF3H1 的活性口袋,与丙氨酸 123(ALA123)、缬氨酸 124(VAL124)形成疏水作用,同时与缬氨酸 124(VAL124)、谷氨酸 122(GLU122)、精氨酸 21(ARG21)、天冬氨酸 330(ASP330)形成氢键(图 5a)。五羟基黄烷酮同样可深入AgF3H1 的活性口袋,与丙氨酸 334(ALA334)、亮氨酸 333(LEU333)形成疏水作用,与天冬氨酸 330(ASP330)、谷氨酰胺 276(GLN276)形成氢键,还能与赖氨酸 215(LYS215)形成 π- 阳离子键(图 5b)。
- 五羟基黄烷酮、柚皮素与AgF3H2 的对接分数分别为 - 6.747 和 - 6.025,MM-GBSA 结合自由能分别为 - 35.60 kcal/mol 和 - 30.49 kcal/mol,表明二者与AgF3H2 的结合稳定性良好。柚皮素可深入AgF3H2 的活性口袋,与苯丙氨酸 319(PHE319)、酪氨酸 323(TYR323)、脯氨酸 220(PRO220)、亮氨酸 214(LEU214)形成疏水作用,与赖氨酸 196(LYS196)、丝氨酸 117(SER117)、精氨酸 128(ARG128)形成氢键(图 5c)。五羟基黄烷酮可深入AgF3H2 的活性口袋,与脯氨酸 220(PRO220)、酪氨酸 323(TYR323)形成疏水作用,与酪氨酸 323(TYR323)、赖氨酸 215(LYS215)形成氢键,还能与天冬氨酸 330(ASP330)、精氨酸 128(ARG128)形成额外氢键,并与组氨酸 217(HIP217)形成 π- 阳离子键与 π-π 键(图 5d)。
综上,显齿蛇葡萄中的AgF3H1 与AgF3H2 基因均更倾向于与五羟基黄烷酮结合;且结合后,AgF3H1 与五羟基黄烷酮的结合稳定性显著高于 AgF3H2 。结合 WGCNA 分析结果,本研究推测:AgF3H1 是催化五羟基黄烷酮合成二氢杨梅素的关键酶基因 。
AgF3H1 基因定位于显齿蛇葡萄的 9 号染色体上,物种间共线性分析结果显示,该染色体在演化过程中发生了基因重排事件(图 3c)。局部共线性分析结果表明,显齿蛇葡萄的AgF3H1 基因,与葡萄属物种 4 号染色体及圆叶白粉藤 5 号染色体上的F3H 基因存在共线性关系(图 5e),这与物种间共线性分析结果一致(图 3c)。实时荧光定量 PCR 实验结果显示,AgF3H1 基因在不同组织中的表达水平与转录组数据高度吻合(图 5f)。
综合以上所有实验结果,本研究最终证实:AgF3H1 是调控显齿蛇葡萄二氢杨梅素生物合成的核心关键基因 。
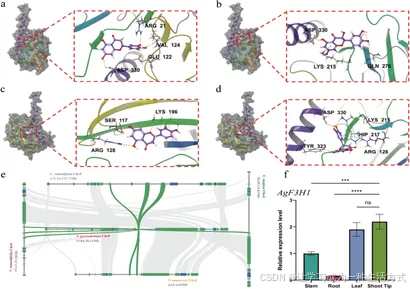
图 5 显齿蛇葡萄 AgF3H1 与 AgF3H2 基因的生物信息学分析
(a) AgF3H1 与圣草酚的分子对接模型;(b) AgF3H1 与五羟基黄烷酮的分子对接模型;(c) AgF3H2 与柚皮素的分子对接模型;(d) AgF3H2 与五羟基黄烷酮的分子对接模型。黄色线条代表氢键;绿色线条代表 π- 阳离子键;蓝色线条代表 π-π 键。(e) 显齿蛇葡萄 9 号染色体与欧洲葡萄、山葡萄、圆叶葡萄、圆叶白粉藤及柑橘对应染色体的局部共线性分析,绿色区域代表AgF3H1 的同源序列。(f) 显齿蛇葡萄不同组织中AgF3H1 基因的实时荧光定量 PCR 分析。数据以三次独立重复实验的平均值 ± 标准差 表示;采用 Student's t 检验进行显著性分析:*P < 0.001 ,**P < 0.0001 ;n.s. 表示无显著差异。
4 讨论
显齿蛇葡萄是我国特有物种,其嫩茎与叶片富含黄酮类化合物,广泛应用于功能性保健饮品及民间药材开发。本研究整合 Illumina 短读长测序、PacBio 长读长测序与高通量 Hi-C 染色体构象捕获技术,成功组装得到显齿蛇葡萄近乎完整的参考基因组。该基因组的scaffold N50 值达 30.45 Mb,contig N50 值达 21.93 Mb ,显著优于其他葡萄科物种(如三叶崖爬藤的 contig N50 为 2.15 Mb、scaffold N50 为 86 Mb)(Zhu 等,2023)及药用植物团花树(contig N50 为 0.82 Mb、scaffold N50 为 29.20 Mb)(Zhao 等,2022)。
最终校正后的显齿蛇葡萄基因组大小为 555.42 Mb,挂载至 20 条伪染色体上,是蛇葡萄属首个近乎完整的参考基因组 。其基因组大小与其他葡萄科物种相近,如山葡萄(约 522.28 Mb)(Wang 等,2024)、山葡萄(604.56 Mb)(Wang 等,2021)、欧洲葡萄(494.87 Mb)(Shi 等,2023)及圆叶葡萄(413.91 Mb)(Huff 等,2023)。
显齿蛇葡萄基因组的杂合度高达 1.48%,重复序列占比达 62.62%,均高于浙江葡萄(杂合度 0.845%、重复序列占比 47.49%)(Li H. Y. 等,2024)、山葡萄(杂合度 1.20%、重复序列占比 59.21%)(Wang 等,2024)及圆叶白粉藤(杂合度 1.19%、重复序列占比 47.41%)(Xin 等,2022)等葡萄科物种。该基因组的短读长比对覆盖率达 99.29%,BUSCO 评估的基因组完整度达 98.80%,优于山葡萄(BUSCO 完整度 97.50%)(Wang 等,2024)、山葡萄(短读长比对覆盖率 98.58%、BUSCO 完整度 94.60%)(Wang 等,2021)及欧洲葡萄(BUSCO 完整度 98.50%)(Shi 等,2023)。上述结果表明,本研究组装的显齿蛇葡萄基因组在序列一致性、组装准确性与完整性方面均优于其他葡萄科植物,为后续系统发育分析、基因功能验证及分子育种研究奠定了坚实基础。
在植物演化历程中,多数物种都经历过一次或多次古老的基因组多倍化事件(Blanc 和 Wolfe, 2004;Jiao 等,2011;Soltis 和 Soltis, 2016)。全基因组复制(WGD)事件对植物物种形成及优良性状的演化具有关键推动作用(Rensing, 2014;Song 等,2024)。由于葡萄属乃至葡萄科植物与被子植物共同祖先的亲缘关系较近,其基因组数据被广泛应用于演化分析研究(Xin 等,2022)。
系统发育分析结果显示,白粉藤属与葡萄属的分化时间约为 49.0 百万年前(置信区间:31.4--69.1 百万年前),这一结果与基于全基因组数据估算的圆叶白粉藤(68.41 百万年前,置信区间 44.1--89.8 百万年前)、翅茎白粉藤(置信区间 60.19--84.68 百万年前)与葡萄属的分化时间基本一致(Xin 等,2022;Li Q. Y. 等,2024);但也有研究表明,白粉藤属与葡萄属的分化时间可能为 38.07 百万年前(置信区间 21.38--67.28 百万年前)(Li 等,2024)。由此可见,本研究获得的高质量显齿蛇葡萄基因组数据,为厘清葡萄科物种间的演化关系与系统发育地位提供了有力支撑。
Ks 与 4DTv 分布图谱呈现的两个峰值表明,显齿蛇葡萄在演化过程中不仅经历了被子植物共有的全基因组三倍化事件(WGT-γ 事件),还发生了一次葡萄科特有的全基因组复制事件(Jaillon 等,2007;Tang 等,2008)。全基因组复制事件不仅会使基因组大小加倍,还会驱动基因拷贝的获得与丢失(Van de Peer 等,2009),进而对植物生长发育及代谢通路调控产生深远影响(Chae 等,2014;Chakraborty, 2018)。
共线性分析结果显示,葡萄科物种间的染色体共线性模式较为复杂;显齿蛇葡萄的 9 号染色体在演化过程中发生了染色体重排及基因片段重排事件。两次全基因组复制事件导致显齿蛇葡萄基因组中大量基因发生扩张、收缩或丢失,祖先基因的同源序列因此分散于多条染色体上。已有研究证实,基因家族扩张是植物适应自然环境变异的核心驱动力,扩张后的基因家族能够显著提升植物对生物胁迫与非生物胁迫的耐受性(Renny-Byfield 和 Wendel, 2014)。本研究发现,显齿蛇葡萄基因组中存在 1114 个扩张的基因家族,这些家族主要富集于次生代谢物生物合成过程 与次生代谢过程 通路。综上,本研究推测:显齿蛇葡萄在经历两次全基因组复制事件及 9 号染色体基因重组事件后,逐步积累了调控黄酮类化合物合成的关键基因,最终形成高黄酮含量的物种特征。
遗传资源挖掘与关键性状候选基因筛选,是解析物种遗传变异规律与环境适应性机制的核心途径。本研究整合基因共表达网络分析与黄酮代谢组分析,系统解析了显齿蛇葡萄黄酮类化合物的生物合成通路及其调控网络。二氢杨梅素是显齿蛇葡萄中含量最高的黄酮单体化合物,其含量变化趋势基本可反映总黄酮含量的变化规律。在显齿蛇葡萄中,AgF3H 与AgF3'5'H 是调控二氢杨梅素合成的核心催化基因,二者可分别催化五羟基黄烷酮、二氢槲皮素及二氢山奈酚生成二氢杨梅素。
本研究利用 WGCNA 分析基因与代谢物的关联性,从黄色、棕色、蓝色及青绿色共表达模块中筛选出 10 个与黄酮合成高度相关的关键基因,包括AgPAL3 、AgPAL2 、AgPAL1 、AgFLS3 、AgFLS2 、AgFLS1 、AgF3H1 、AgDFR 、AgCHS2 及AgCHS1 。转录组测序与 qRT-PCR 验证结果显示,AgF3H1 基因在嫩梢组织中的表达量最高,叶片、茎、根组织中的表达量依次降低,这一表达模式表明AgF3H1 的高表达可能是显齿蛇葡萄二氢杨梅素高含量的关键原因,与前人研究结论一致。
基于近乎完整的基因组序列,本研究在显齿蛇葡萄基因组中鉴定出 2 个F3H 家族基因(AgF3H1 与AgF3H2 )。分子对接分析结果显示,虽然两个AgF3H 基因均对五羟基黄烷酮具有较高的结合亲和力,但AgF3H1 与五羟基黄烷酮结合后的稳定性(结合自由能 - 41.49 kcal/mol)显著高于AgF3H2 (结合自由能 - 30.49 kcal/mol)。已有研究证实,F3H 基因的高表达可显著提升植物组织中的黄酮含量:将番茄SlF3H 基因转入烟草后,过表达植株的黄酮含量较野生型提高约 30%(Meng 等,2015);将茶树CsF3Hs 基因转入拟南芥后,转基因植株种子中多数黄酮醇苷与低聚原花青素的含量显著提升(20%--40%),表明CsF3Hs 在茶树黄酮类化合物合成中发挥核心调控作用(Han 等,2017)。上述研究均证实,F3H 基因的表达调控对植物黄酮代谢与合成具有显著影响,凸显了AgF3H 基因在显齿蛇葡萄黄酮类化合物及其衍生物合成中的关键地位。
因此,结合 WGCNA 分析结果,本研究推测AgF3H1 是催化五羟基黄烷酮合成二氢杨梅素的关键酶基因 。在后续研究中,将通过同源与异源表达系统对AgF3H1 基因进行全面的功能验证,包括在宿主物种中开展过表达分析、在模式生物中进行异源表达验证,以及利用 CRISPR/Cas9 介导的基因编辑技术开展靶向基因敲除实验。通过多平台验证策略,系统解析该基因在黄酮生物合成通路中的调控机制及其对植物生理过程的多效性影响。研究结果将为通过分子育种改良作物次生代谢产物谱奠定理论基础,同时为利用代谢工程技术开发生活性成分强化型农产品提供技术参数。
综上所述,本研究首次完成了显齿蛇葡萄近乎完整的基因组组装,为该物种及其他药食同源植物的深入研究提供了全面的基因组数据支撑;比较基因组演化分析阐明了葡萄科植物的演化轨迹;黄酮合成通路候选基因的挖掘,为显齿蛇葡萄的遗传改良提供了重要靶点。