论文题目:Efficient evolution of human antibodies from general protein language models
人类抗体从通用蛋白质语言模型的高效进化(Brian L. Hie 1,2 , Varun R. Shanker 2,3, Duo Xu 1,2, Theodora U. J. Bruun 1,2,3, Payton A. Weidenbacher 2,4, Shaogeng Tang 1,2, Wesley Wu 5, John E. Pak5 & Peter S. Kim 1,2,5)nature biotechnology
个人总结:
主动学习是:模型主动选择样本-实验标注-反馈-继续训练
这个文章为:使用plm进行一次筛选-做实验验证。
文章主要创新点:使用多个PLM进行筛选出共识高效变异,在验证时,第一轮使用BLI保留有效的突变,第二轮将第一轮优质突变整合看看能否得到多突变组合。
特点:快,不需要知道太多先验细节,实验高效,能在蛋白质家族上通用
这里表达的整个意思就是单一的PLM的可能不准,所以集成了多个模型来推荐。
首先假设一个酶序列X,长度100,第50位xi的为丙氨酸,这里作者用了6个语言模型ESM1-b+ESM1-v来筛选突变
首先每个模型单独工作,把第50位xi的换成AAA酸后这个新氨基酸在该位置的概率,只要这个模型计算出来的概率比野生型xi的概率大 就推荐第50位的位置换成AAA酸。
然后统计多少个模型体检了同一个突变,如果第50位同时被多个模型推荐换为aaa 他就会被推荐出来。
摘要
- 自然进化必须探索广阔的序列,以发现理想但罕见的突变,这表明从自然进化策略中学习可以指导人工进化(这和进化计算差不多)。
- 本文报告,通用的蛋白质语言模型可以通过提出进化上合理的突变,高效地进化人类抗体,尽管模型未提供任何关于目标抗原、结合特异性或蛋白质结构的信息。
- 对七个抗体进行了语言模型引导亲和力成熟,筛查每种抗体的20个或更少变异体,仅在两轮实验室演化中筛选,并将四个临床相关、高度成熟抗体的结合亲和力提升至七倍,三个未成熟抗体的结合亲和力提升至160倍,许多设计还显示出对埃博拉和严重急性呼吸综合征冠状病毒2型(SARS-CoV-2)的良好热稳定性和病毒中和活性伪病毒。提升抗体结合的模型同样指导着不同蛋白家族和选择压力(包括抗生素耐药性和酶活性)的高效进化,表明这些结果可推广到多种情境。
引言
进化在浩瀚的可能序列中寻找能提升适应度的罕见突变。在自然界中,这种搜索基于简单的随机突变和重组过程,但在实验室中用同样的方法进行蛋白质定向进化则带来了相当大的实验负担。基于随机猜测或暴力搜索的人工进化通常投入大量精力去探究弱活性或无功能的蛋白质,需要高实验通量以识别适应度更好的变异。
虽然进化适应度部分由特定的选择压力决定,但也存在一些更普遍适用于蛋白质家族的特性 ,或是大多数蛋白质适应度和功能的前提条件 ;例如,某些突变能维持或改善 稳定性或进化性,而另一些突变则会破坏结构稳定性或诱导出无能、错误折叠的状态。提高进化效率的一种方法是确保突变遵循这些我们称之为进化合理性的一般性质。识别合理的突变有助于引导进化远离无效的系统,从而间接提升进化效率,而无需明确了解相关功能。
然而,这一策略也具有挑战性,首先,蛋白质序列受复杂规则支配 ;其次,即使我们将搜索限制在进化上合理的突变中,能够改善特定适应度定义的突变可能仍然稀少,超出实际实用价值(见图1a,突变合理,但是有用的突变比较少)。更广泛地说,一个主要的未解问题是,一般进化信息(例如,从过去进化中获得的序列变异学习模式)是否足以在特定选择压力(例如对特定抗原的更高结合亲和力)下实现高效进化。
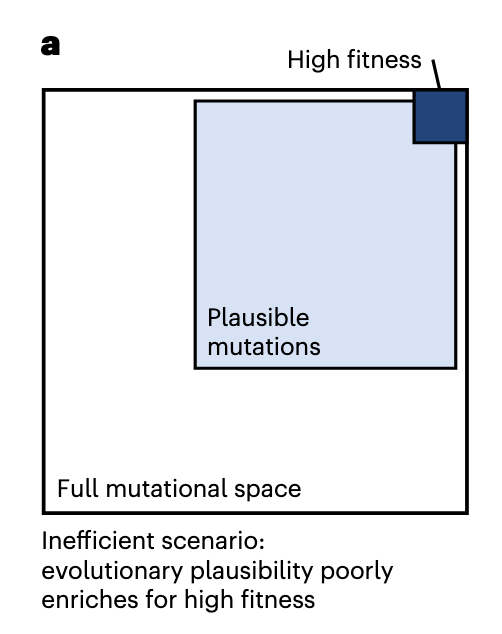
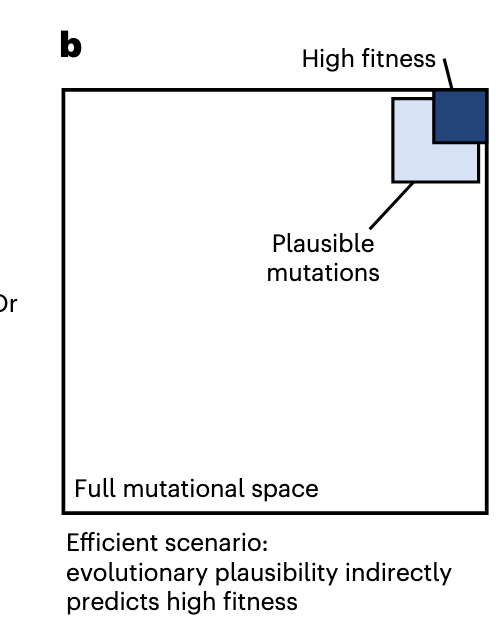
我们展示了仅靠进化信息就能在特定选择压力下高效提升适应度(见图1b,突变合理且有用的也非常多)。在我们的主要实验案例中,我们关注人类抗体的亲和力成熟(亲和力成熟本质是让生物分子(比如酶、抗体)对目标分子(酶的底物、抗体的抗原)的 "结合能力" 变得更强的优化过程 ),其中特异性选择压力定义为对特定抗原更强的结合亲和力。在自然界中,一种称为体细胞超突变 的过程通过反复诱变,使抗体谱系进化或"成熟",使其对抗原更具亲和力。在实验室中,亲和力成熟是定向进化的重要应用,因为具有高疾病亲和力的抗体具有治疗潜力。
为了选择进化上合理的突变,我们使用了称为语言模型的算法(见图1c)来学习可能出现在天然蛋白质中的模式 。由于我们使用了通用语言模型,训练于非冗余序列数据集 ,这些序列数据集旨在代表所有天然蛋白的变异,这些模型只能学习比专门训练在抗体序列上训练的模型更一般的进化规则,或直接由结合亲和力监督的模型。给定一个起始序列,我们利用这些语言模型推荐合理的氨基酸替代,并对其进行实验筛查以提升适应度。对最终用户来说,该算法只需一条野生型序列,无需初始结合亲和力数据、抗原知识、任务特异性监督、进化同源物或蛋白质结构信息。
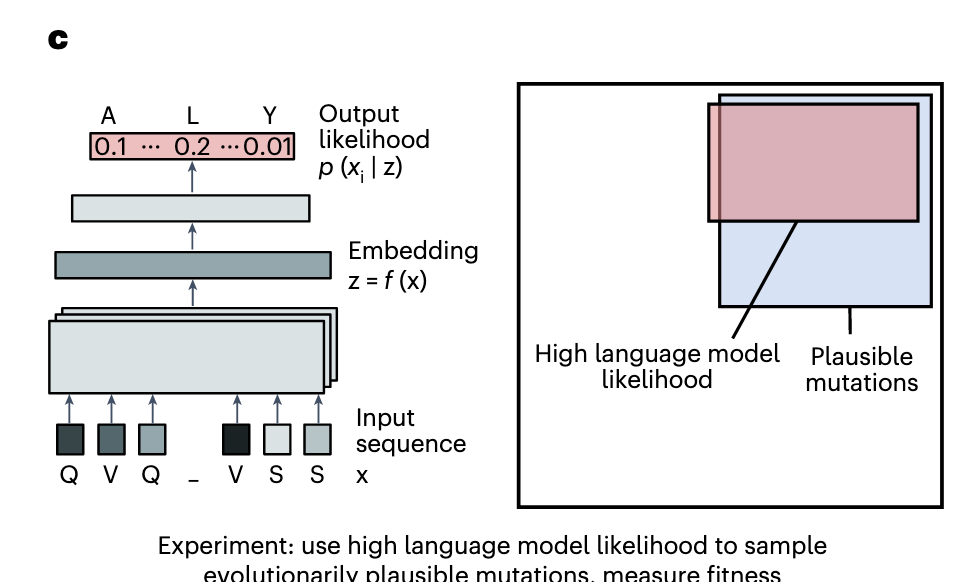
这个展示了PLM的全流程 包括输入(氨基酸)然后特征嵌入(把氨基酸转化为高维特征向量)在给不同的氨基酸打分  这里表示自然界中位置更常出现L ,L是相对来说进化合理的。
这里表示自然界中位置更常出现L ,L是相对来说进化合理的。
所以相比于图a,图b PLM可以帮助筛选出合理的突变。
结果
A.蛋白质语言模型的高效亲和力成熟
最新研究表明,尽管语言模型对特定选择一无所知,也能预测自然进化。然而,这项先前的工作只有在对进化轨迹的全面了解后,才回溯性地预测进化方向 。我们假设蛋白质语言模型的预测能力可能使研究人员仅向算法提供单一野生型抗体序列 ,并接收一小组可管理的高似然变异(~101)以实验测量其理想性质 。这是一个非常通用的设定,不假设你了解蛋白质结构或任务特定的训练数据。然而,一个重要问题是,更高的进化概率是否能有效地转化为更高的适应度。我们通过进行进化运动,基于语言模型似然度,测试代表不同抗原和成熟度的七种抗体亲和力成熟
我们使用ESM-1b语言模型和ESM-1v五个语言模型(共六个语言模型)进行了进化。ESM-1b和ESM-1v分别在UniRef50和UniRef90上训练,这两组蛋白质序列数据集代表了数百万自然蛋白的变异(UniRef90包含~980万条总序列),仅包含几千个抗体相关序列23。这些数据集还设计得确保序列间的序列相似度不超过50%(UniRef50)或90%(UniRef90),以避免生物学上的冗余。此外,这两个数据集都早于研究中SARS-CoV-2抗体的发现以及所有相关SARS-CoV-2变异株的演化。因此,为了进化这些抗体,语言模型不能在训练数据中使用疾病特异性偏差,必须学习更通用的进化模式。
我们利用这些语言模型计算了所有单残基置换对重链(VH)或轻链(VL)抗体变量区域的概率。我们在六个语言模型共识中选择了进化概率高于野生型的替换(方法与扩展数据图1)。在第一轮进化中,我们通过生物层干涉测量(BLI)对仅含有野生型单残基取代的变异体的抗原相互作用强度。第二轮中,我们测量了包含替换组合的变异,并根据第一轮结果选择了对应保留或改善结合的替换。我们对所有七个抗体进行了这两轮,第一轮每个抗体检测到8--14个变异体,第二轮每个抗体检测到1--11个变异体(见图2及补充表1)。临床相关抗体的变异体,这些变异体的解离率极低或无法检测为IgG,通过测量单价片段抗原结合区(Fab)的解离常数(Kd)进行筛选;通过测量二价IgG的表观Kd,随后测量高亲密度变异的Fab片段的Kd值,筛选未成熟抗体的变异株(方法)
我们能够成功表达122个变异中除一个外的所有,贯穿我们的七个进化轨迹。在所有七种抗体中,我们发现71--100%的第一轮Fab变异(含单残基取代)保留了亚微粒摩尔对抗原的结合,14--71%的第一轮变体导致结合亲和力提升(定义为Kd比野生型提升1.1倍或更高)(补充表1)。大多数第二轮变体(包含多种替换组合)也改进了绑定(补充表1--9)。除REGN10987抗体外,我们也获得了Kd至少提升两倍的变异。在所有76种语言模型推荐的单残基替换中,有36种(以及32种替代中18种导致亲和力改善)发生在框架区域(补充表2--9),而这些区域在传统亲和力成熟过程中通常比互补性决定区域(CDRs)更少突变12。
尽管这些抗体已高度进化(起始于低纳摩尔或低皮科莫尔亲和力),我们仍成功提升了所有临床相关抗体的结合亲和力。MEDI8852是一种强效结合剂,具有亚皮科莫尔的Fab Kd,覆盖多种HAs,并且能与H4和H7亚型的皮科莫拉或纳摩尔结合。虽然我们明确筛查了使用HA H4抗原的变异株,但最佳设计还能改善广泛HAs的结合能力(补充表2和3),包括HA H7 HK17(A/香港/125/2017(H7N9))的结合率提升了七倍(从0.21 nM提升至0.03 nM)。mAb114的最佳变异体,作为一种临床批准药物,在埃博拉病毒GP的法式Kn表现提升了3.4倍(补充表5)。REGN10987,亲和力最高的变异株通过六个稳定脯氨酸取代(S-6P)40(筛查用抗原)对Beta变异株Spike提升了1.3倍,我们的另一项设计对Omicron BA.1受体结合结构域(RBD)提升了5.1倍(补充表8)。对于S309,我们将设计与野生型及VH中引入N55Q替代的变异进行了比较,经过小规模理性进化筛选35;S309 Fab 与 VH N55Q 取代后形成治疗抗体索托维单抗的 Fab。我们最好的S309变异株亲和力高于索托维单抗,包括在SARS-CoV-2武汉-胡-1 S-6P(筛查用抗原)中,Fab Kd比野生型S309提升1.3倍(索托维单抗为1.1倍);Beta S-6P的提升为1.7倍(索托维单抗为1.3倍);Omicron RBD的变化为0.93倍(索托维单抗为0.82倍)(补充表7)。
我们还改善了三种未成熟抗体的亲和力,且其倍数变化通常远高于成熟抗体,表明亲和力的进化性更为明显。对于MEDI8852 UCA,最佳Fab设计在对HA H1所罗门(A/所罗门群岛/2006/3/2006(H1N1))的抗原(筛查中)的Kd提升了2.6倍。我们的最佳设计还能获得部分二级肝菌的结合宽度,包括湖北HA H4(A/swine/Hubei/06/2009(H4N1))的结合提升了23倍,HA H7 HK17的结合力提升了5.4倍(补充表4)。对于mAb114 UCA,我们的最佳Fab设计使埃博拉病毒GP的Kd提升了160倍(补充表6)。虽然算法建议对这两种UCA抗体进行氨基酸替换,且这些氨基酸置换在成熟抗体中也存在,但成熟版本中并未发现其他增强亲和力的置换:排除成熟抗体中发现的任何替换或修饰位点,我们的UCA变异体在HA H4湖北(变异体VH P75R/VL G95P;补充表4)以及埃博拉病毒GP(变异型VH G88E/VL V43A)的改进了33倍;补充表6),证明我们的算法成功探索了替代进化路径。对于C143,一种在广泛亲和力成熟前分离的患者来源抗体38,39,我们的最佳设计使β-S-6P提升了13倍,Omicron RBD提升了3.8倍(补充表9)。我们定向进化活动的结果在图2、补充表2--9和补充数据1中进一步总结。
B.进化抗体的进一步表征
虽然我们明确选择了改进的结合剂,但也测试了这些变体以提升稳定性(方法)。我们发现Fabs Our进化出的mAb114、mAb114 UCA、REGN10987和C143变异体也能保留或改善Tm;我们观察到的最大变化是mAb114 UCA演化时气温从74.5°C升至82.5°C。然而,热稳定性的提升并不能完全解释亲和力成熟的结果,因为我们观察到亲和力成熟的MEDI8852及其UCA变体的Tm略有下降,尽管这些Fabs仍然具有热稳定性(见图2)。
此外,我们测试了亲和成熟设计中的多特异性结合,因为结合非预期靶点在治疗环境中可能导致不良副作用。对于七种抗体中的每一种,我们都使用多特异性测定法测试野生型及三种亲和成熟变异体,该测试法评估其与可溶性膜蛋白的非特异性结合能力(方法)41,42。我们观察到七种抗体中任何变异的多特异性均无显著变化,所有测试抗体的多特异性值均在可治疗范围内(图3a及补充数据2)。另一个治疗考虑是免疫原性。尽管免疫原性预测仍具挑战性,尤其是识别不连续表位,但线性肽的免疫原性已被更好地理解43。我们观察到,亲和成熟变异株在计算预测的肽结合蛋白结合剂数量上无显著增加(单侧二项P>为0.05),该肽结合剂数量涵盖人类白细胞抗原(HLA)I类和II类(HLA类抗原的确切P值和样本量见补充数据2),这支撑了T细胞介导的免疫原性。
我们还想确定亲和成熟的变异株是否具有更好的病毒中和活性。我们使用伪病毒中和分析方法测试了四个抗体的亲和力增强变异株,并在所有案例中观察到抑制浓度(IC50)值为半最大且显著改善的变异株(Bonferroni校正的单侧t检验P < 0.05,n=4个独立实验),其中对埃博拉伪病毒的最佳mAb114变异株提升了1.5倍;对抗SARS-CoV-2 β假病毒的最佳REGN10987变异株实现了两倍提升;而对抗β伪病毒的最佳C143变异株则提升了32倍(图3b,扩展数据图2及补充表5、8和9)。此外,亲和成熟的mAb114 UCA变体在比野生型低>100倍的浓度下表现出可检测的中和效果(扩展数据图2a)。一般而言,结合亲和力的变化与中和变化良好相关(Spearman r = 0.82,双侧t分布P = 1.9×10⁻⁻,n=15个抗体变体)(图3c及扩展数据图2b)。
C提升亲和力替代的原创性
虽然找到亲和力的任何改进本身对工程应用很有用,但我们也关心算法推荐的一些改动是否体现了"原创性"。我们通过计算某一残基在自然界中被观察到的频率来量化原创性(方法)。尽管在模型的训练数据和抗体序列数据库中确实观察到许多增强亲和力的替换以高频率出现44,但其他替换则展现出更高的原创性。例如,在MEDI8852 UCA的轨迹中,VL G95P框架替代(见图2及补充表4)涉及将99%天然抗体序列中观察到的甘氨酸转变为少于1%的自然序列中观察到的脯氨酸。总体而言,32种增强亲和力的替换中有5种(~16%)涉及将野生型残基转变为罕见或罕见的残基(补充表10),且在仅考虑同一生殖系基因衍生抗体的自然变异时,这种情况也较为罕见(补充表11)。这些结果表明,语言模型既学习涉及高频残基的"简单"进化规则,也学习多序列比对或传统抗体进化无法捕捉的复杂规则。从概念上讲,这些低频、增强亲和力的替换类似于其他学科中的例子,人工智能程序偶尔做出不寻常但有利的选择(例如,不直观的游戏决策45),同样可能更具价值。提升亲和力的替代的原创性 虽然找到亲和力任何改进的能力本身对工程应用有用,但我们也关注一些变化是否存在我们的算法推荐,展示"原创性"。我们通过计算某一残基在自然界中被观察到的频率来量化原创性(方法)。尽管在模型的训练数据和抗体序列数据库中确实观察到许多增强亲和力的替换以高频率出现44,但其他替换则展现出更高的原创性。例如,在MEDI8852 UCA的轨迹中,VL G95P框架替代(见图2及补充表4)涉及将99%天然抗体序列中观察到的甘氨酸转变为少于1%的自然序列中观察到的脯氨酸。总体而言,32种增强亲和力的替换中有5种(~16%)涉及将野生型残基转变为罕见或罕见的残基(补充表10),且在仅考虑同一生殖系基因衍生抗体的自然变异时,这种情况也较为罕见(补充表11)。这些结果表明,语言模型既学习涉及高频残基的"简单"进化规则,也学习多序列比对或传统抗体进化无法捕捉的复杂规则。从概念上讲,这些低频、增强亲和力的替换类似于其他学科中人工智能程序偶尔做出不寻常但有利选择的例子(例如,不直观的游戏决策45),同样值得进一步研究。
剩下的看不懂先略过
讨论
我们证明,仅凭野生型抗体序列,通用的蛋白质语言模型就能指导高效的亲和力成熟。尽管我们的亲和力提升低于通常在体内成功进化轨迹中观察到的,体细胞超突变探索的突变空间大了数个数量级(扩展数据图7)。此外,我们对未成熟抗体的亲和力提升达到了2.3倍至580倍的范围,此前该系统采用先进的体外进化系统应用于未成熟抗RBD纳米抗体(在该系统中,计算部分只需数秒时间,但被持续数周的细胞培养和分选轮次取代)14(扩展数据图7)。体外细胞表面展示方法也面临物理限制,使得当野生型结合剂已有较高亲和力(<1 nM)5时,区分更优结合剂具有挑战性,这并非我们方法的局限。
更广泛地说,我们研究的一个关键发现是,单靠进化信息在选择少量替代以测试适应度提升时,提供了足够的先验信息(见图1b和4b)。这导致了一个没有任务特异性训练数据或抗原知识的模型,能够引导抗体进化达到更高的结合亲和力,且与蛋白质特异性或任务特异性方法相比,其性能具有竞争力(补充表12及扩展数据图5)。我们假设,在许多环境中,当突变被限制遵循一套一般进化规则时,相当一部分(超过10%)必然会改善适应度(见图4b),这对实验室和自然界的进化具有直接且更广泛的影响。
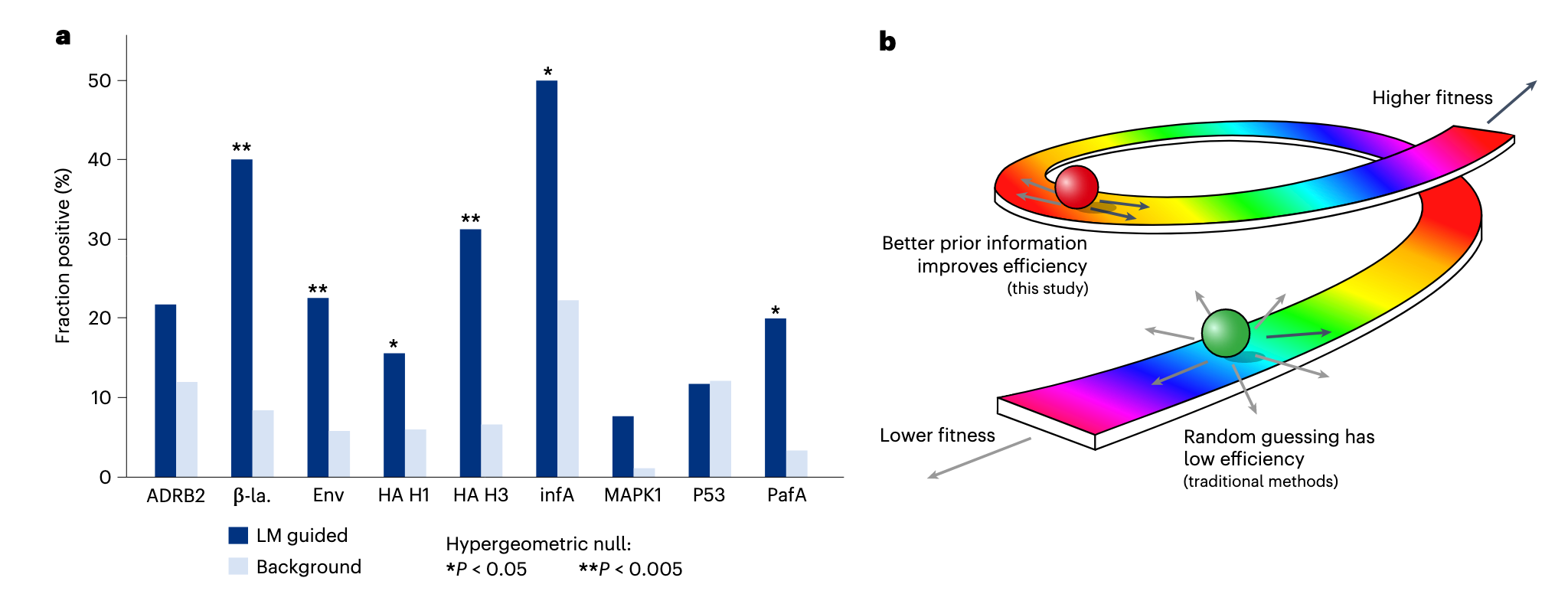
相比于随机突变,ML指导的突变更有效果,这证明了PLM不需要你想要提升酶的什么性能,他只推荐在他的计算下比较有用的推荐,相比于随机变化这已经有很大的进步。
适应度改进的普遍性
通过利用一般进化规则,语言模型推荐更"普遍"的变化,当适应度定义改变时,这些变化似乎更能推广(见图4)。我们还观察到一般语言模型优于抗体特异性语言模型(补充表12),这与独立的计算机基准测试结果一致22。当转向新的、特定的适应度概念时,更一般的进化信息可能超过抗体库数据集中编码的特定偏倚,尽管进一步开发抗体语言模型有望提升性能。我们的总体方法是改进现有的基线功能(例如,提高弱结合剂的亲和力),而不是赋予任何蛋白质任意功能(例如将通用蛋白转化为特定结合剂)。我们还注意到,当选择压力不自然或野生型序列已达到适应度峰值时,利用这种策略引导进化可能更为困难。然而,在许多实际设计任务中,自然序列和选择压力已经是更优选择的;例如,治疗开发通常会优先考虑人类抗体,因为考虑了免疫原性。除了蛋白质工程,我们方法的成功也可能为自然进化提供见解。进化信息的效率本身可能反映了自然机制,使突变率偏向更高适应度:例如,体细胞超突变通过表观基因组和酶序偏倚偏向抗体基因的特定部分60,61。如果表观基因组或其他机制使突变具有高适应度,那么自然界可能正以类似我们方法的方式加速进化。
方法
通过语言模型共识获取氨基酸替换
我们选择语言模型共识推荐的氨基酸替换。我们以单一野生类型序列x = (x1,...,xN)∈XXN为输入,其中X是氨基酸集合,N是序列长度。我们还需要一组掩蔽语言模型,这些模型经过预训练产生条件似然p(x′|x)。为了指导基于某个语言模型的进化,我们首先计算语言模型似然高于野生类型的替换集合------即计算集合
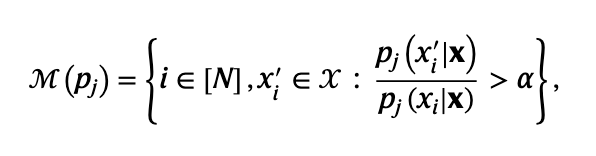
其中 pj 表示语言模型,xi表示野生型残余,且 α = 1。为了进一步筛选替换,仅筛选出最有可能的替换,我们选择基于共识方案的替换,其中对于新的氨基酸 xi′,我们计算
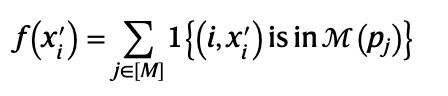
其中1{·}表示指示函数,存在M个语言模型。然后我们获得在多个语言模型中比野型更可能的替换集合------即我们获得
其中k是用户提供的截止值,控制对应变异的数量。虽然我们关注的是k中产生小值的|AA|(约10)可通过低通量检测筛查,但通过降低k的值或降低截断紧度α,可以增加替换次数。我们的策略基于计算基于整个序列p(习′|x)的"野生型边际"似然,而非被掩蔽的"掩蔽边际"似然p(习′|xN\{i}),也增加了截断紧密度(扩展数据图1)。我们使用六个大规模掩码语言模型------即ESM-1b model19和五个被集成成ESM-1v20的模型------均来自 https://github.com/facebookresearch/esm。ESM-1b基于2018-03年UniRef50(参考文献23)训练,包含约2700万序列,ESM-1v中的五个模型分别基于2020-03年发布的UniRef90(参考文献23),共约9800万序列。
这里表达的整个意思就是单一的PLM的可能不准,所以集成了多个模型来推荐。
首先假设一个酶序列X,长度100,第50位xi的为丙氨酸,这里作者用了6个语言模型ESM1-b+ESM1-v来筛选突变
首先每个模型单独工作,把第50位xi的换成AAA酸后这个新氨基酸在该位置的概率,只要这个模型计算出来的概率比野生型xi的概率大 就推荐第50位的位置换成AAA酸。
然后统计多少个模型体检了同一个突变,如果第50位同时被多个模型推荐换为aaa 他就会被推荐出来。
抗体序列分析与演化
对于抗体,我们分别对VH和VL序列执行上述步骤,分别获得AAVH和AAVL组。在进化的第一轮中,我们设定α = 1,并选择k的值,使得|AAVH与AAVL的 ∪约为10,这相当于一个人可以并行表达和纯化的合理数量的抗体变异。我们使用k = 2表示MEDI8852 VH和VL,k = 2表示MEDI8852 UCA VH和VL,k = 4表示mAb114 VH和VL,k = 2表示mAb114 UCA VH和VL,k = 2表示S309 VH,k = 1表示S309 VL,k = 1表示S309 VL,k = 2表示REGN10987 VH和VL,k = 2表示C143 VH和VL。我们进一步缩小了|的大小AAVH∪AAVL|要求替换在至少一个语言模型中,其位点具有最高概率。首先通过BLI测量对某抗原的结合亲和力(详见下文),增强亲和力的变异被重组,使得第二轮变异具有野生型的两个或以上替换,这些替换在进化的第二轮中进行了测试。鉴于S309和REGN10987在进化第一轮中发现的增强亲和力的替换数量较少,我们还扩展了第二轮考虑的替换集,包括那些保持亲和力的替换。对于MEDI8852和MEDI8852 UCA,我们在第二轮测试了所有可能的组合;对于其他可能组合远超~10个变异的抗体,我们手动选择一组组合,优先纳入在第一轮中亲和力提升最大的替代。我们使用了原研究作者提供的野生型序列,描述了各自抗体29--32,38。野生型VH和VL序列见补充信息。我们使用abYsis webtool版本3.4.1(http://www.abysis.org/abysis/index.html)44提供的Kabat区域定义,注释了VH和VL序列中的框架区域和CDR。
抗体亲和度基准实验
我们还比较了上述策略推荐的替换(基于语言模型共识)与四种基于序列的替代方法推荐的替换。首先,我们基于位点无关的突变频率对VH或VL序列进行了替换,其中我们使用了abYsis Annotation webtool44计算的频率,或是UniRef90(ESM-1v的训练数据集)中所有序列获得的频率23。为了计算UniRef90频率,我们首先进行了穷尽搜索,以Levenshtein距离获得10,000条最接近的序列,其中10,000条代表UniRef90中免疫球蛋白样序列的数量。我们使用FuzzyWuzzy Python 0.18.0版本中的partial_ratio函数计算序列相似性;随后,我们使用MAFFT版本7.475(参考文献63),以VH或VL序列为参考,构建了这1万个序列的多重序列比对;最后,利用比对计算序列中每个位点的突变频率。我们通过似然比(突变频率除以对应的野生型频率)选择了VH和VL序列中排名最高的替换,每种抗体选择了与进化活动第一轮考虑的相同数量的替换。
我们还基于专门训练于抗体序列的语言模型获得了替换。我们使用AbLang重链和轻链语言模型(https://github.com/ TobiasHeOl/AbLang)24以及Sapiens重链和轻链语言模型(https://github.com/Merck/Sapiens)25,计算了所有单残基置换对VH或VL序列的突变型与野生型似然比(使用对应链序列训练的语言模型)。我们根据VH和VL序列的似然比选择了排名最高的替换,并根据通用蛋白质语言模型的使用,也要求置换在其位点具有最高似然。对于每个抗体,我们选择了与进化研究第一轮考虑的相同数量的替代。我们使用这四种方法(abYsis、UniRef90、AbLang和Sapiens)选择三种未成熟抗体(MEDI8852 UCA、mAb114 UCA和C143)的替换,并用BLI测量IgG对其抗原(HA H1 Solomon、GP和Beta S-6P)的亲和性。为了纯化这些基准研究中更多的变异,我们采用了中通量系统和机器人液体处理机,详见下文。通过该系统,我们表达并纯化了含有野生型单残基置换的抗体变异,这些变异由ESM语言模型共识推荐,以及四种基线方法,观察到相同变体在通过我们进化研究中使用的低通量系统(下文所述)表达和纯化时,其纯度和亲和力相似。中通量纯化后,200微升中最终浓度低于0.1 mg ml−1的抗体,采用低通量方法重新表达和纯化。
UniRef90 鲁棒性和统计显著性分析
这里就是做实验验证了。