随着冷冻电子显微镜(Cryo-EM)技术的飞速发展,"分辨率革命"使得近原子乃至原子级分辨率的生物大分子结构成为常态。如今,大量高价值靶点(如膜蛋白、大型复合物、柔性机器)的结构首次通过Cryo-EM解析,并迅速被用于药物设计、功能机制研究和分子动力学(MD)模拟。截止2025年12月,RCSB PDB数据库中记录了3.2万条通过Cryo-EM获得的晶体结构,仅次于X-ray结构的数量,最优分辨率已接近1 Å。
然而,与X射线晶体学不同,Cryo-EM结构具有独特的数据特征和建模挑战------局部分辨率不均、建模不确定性高、可能存在过度拟合。若未经严格评估就直接用于MD模拟,极可能导致模拟从错误构象出发,不仅浪费宝贵的计算资源,更可能得出误导性结论。
本文系统梳理Cryo-EM结构用于MD前必须完成的质量评估框架与预处理流程,帮助研究者科学判断结构可靠性,并做出合理决策。
一、为什么Cryo-EM结构需要特殊评估?
Cryo-EM的工作流程决定了其结构的独特性:
- 样品状态:蛋白质被快速冷冻在玻璃态冰中,更接近生理环境;
- 数据采集:从数万至数百万张二维投影图像重构三维密度图;
- 建模方式:将原子模型"拟合"进电子密度图,而非直接从衍射数据推导。
这一过程引入了几个关键问题:
- 局部分辨率异质性:刚性区域(如催化核心)分辨率高,柔性区域(如loop、末端)模糊甚至缺失;
- 建模主观性:在中低分辨率区域,建模者可能"脑补"侧链或主链走向;
- 过度拟合风险:为追求高几何分数,模型可能强行匹配噪声;
- 氢原子缺失:除非分辨率<1.8 Å,否则PDB中通常不含氢,影响质子化状态判断。
因此,"高全局分辨率 ≠ 高质量模型"。必须结合多维指标进行综合评估。
二、核心评估维度与关键指标
1. 全局与局部分辨率:可靠性的基石
全局分辨率 是首要筛选标准,通常以FSC = 0.143截断值报告(如"3.2 Å")。其意义如下:
| 分辨率范围 | 原子级信息可靠性 | MD适用性建议 |
|---|---|---|
| < 2.5 Å | 侧链清晰、可分辨水分子、部分氢可见 | 理想,可全原子MD |
| 2.5--3.5 Å | 主链可靠,大部分侧链可建模 | 适合功能位点MD,需验证侧链 |
| 3.5--4.5 Å | 主链可信,侧链多为"棒状"建模 | 谨慎使用,建议约束或简化侧链 |
| > 4.5 Å | 仅Cα骨架可靠,侧链高度不确定 | 不推荐全原子MD,考虑粗粒化 |
注意:即使全局分辨率为3.0 Å,若活性位点局部分辨率>4.0 Å,则该区域不可信。
局部分辨率图揭示了结构内部的质量差异。工具推荐:
- ChimeraX :
Volume Viewer → Local Resolution; - ResMap 或 blocres(命令行)。
评估要点:
- 功能关键区(配体结合口袋、催化残基、变构位点)是否≤3.0 Å?
- 柔性loop是否被过度建模(如在5 Å区域画出完整侧链)?
- 跨膜区或蛋白-蛋白界面是否足够清晰?
2. 模型--密度拟合质量:避免"空中楼阁"
这是最关键的一步。再高的分辨率,若模型未真实反映密度,也是无效的。
(a)可视化检查(必须人工完成)
使用 COOT (首选)或 ChimeraX 加载PDB与MRC密度图:
# ChimeraX 示例
open model.pdb
open emd_12345.map
volume #2 level 0.05 # 调整等值面至EMDB推荐值(通常0.02--0.08)重点检查:
- 侧链取向:芳香族(Phe/Tyr/Trp)、带电残基(Arg/Lys/Asp/Glu)是否"坐"在密度中?有无"飘在空中"?
- 主链走向:是否平滑连续?有无锯齿状异常或悬空羰基?
- 配体/辅因子:是否有连续、形状匹配的球形密度?占有率是否合理?
- 金属离子:是否有高密度峰?配位几何是否合理(如Zn²⁺四面体)?
危险信号:配体密度弱但构象高度扭曲;大量侧链指向无密度区域。
(b)量化指标
- Q-score (Pintilie et al., 2020):衡量单个原子与局部密度的匹配度(0--1)。
>0.7 为优秀,<0.5 表示严重不匹配。
在线计算:https://qscore.emdatabank.org/ - CC_mask (Phenix):模型与掩膜内密度的相关系数。
>0.8 良好,<0.7 需警惕。 - EMRinger score (Phenix/MolProbity):评估芳香族/甲硫氨酸侧链rotamer与密度一致性。
>2.0 优秀,<1.0 表示侧链可能错误。
3. 几何与立体化学质量:防止力场崩溃
即使高分辨率Cryo-EM结构也可能存在建模错误。使用 MolProbity(集成于Phenix/ChimeraX)进行全面验证:
phenix.molprobity model.pdb map.mrc resolution=3.2关键指标与阈值:
| 指标 | 优秀(<3.0 Å) | 可接受(3.0--4.0 Å) | 危险信号 |
|---|---|---|---|
| Ramachandran favored | >98% | >95% | <90% |
| Rotamer outliers | <0.5% | <1% | >2% |
| Clashscore | <5 | <10 | >20 |
- Ramachandran favored:落在 Favored 区域的残基百分比 。在 Ramachandran 图 (拉氏图)中,氨基酸残基的主链二面角(φ, ψ)落在能量有利、立体化学允许的区域 的比例。区域划分 (以 MolProbity 为准):Favored(优势区) :最合理的构象(如 α-螺旋、β-折叠核心区); Allowed(允许区) :可接受,但略偏离理想;Outliers(异常值):构象不合理,可能建模错误
- Rotamer outlier :侧链 χ 角落在罕见或高能区域 (<1% 概率),可能表示建模错误或真实但特殊的相互作用。Rotamer(旋转异构体) :由于单键旋转受限,侧链倾向于采取若干离散的、能量有利的构象 。例如:Val、Thr :通常有 2--3 种常见 rotamer,Arg、Lys :因长链柔性大,rotamer 更多但仍有偏好Phe、Tyr、Trp:芳香环取向受堆积作用约束
- Clashscore:每1000原子中的严重碰撞数。Cryo-EM因含氢,通常比晶体结构更低。
4. 生物合理性:人工智慧的最后一道防线
自动化工具无法替代生物学直觉。需人工判断:
- 氢键/盐桥网络:是否形成合理相互作用?有无孤立带电残基?
- 疏水核心:非极性残基是否被合理埋藏?
- 配体取向:是否符合已知SAR(构效关系)或催化机制?
- 对称性:若为同源寡聚体,各亚基构象是否一致?
三、MD导向的预处理策略
通过评估后,需对结构进行针对性优化,才能安全用于MD。
1. 处理柔性与缺失区域
Cryo-EM常因柔性而缺失loop或末端:
- 短缺失(<5残基) :用 MODELLER 或 Rosetta 补全,并验证是否与局部分辨率图冲突;
- 长缺失/无序区 :不要强行建模!在MD中可设为柔性或直接截断;
- 高B因子区域(>80--100 Ų):考虑删除或施加弱约束。
工具建议 :使用 ISOLDE(ChimeraX插件)进行密度引导的交互式建模,特别适合中低分辨率区域。
2. 质子化状态与力场兼容性
Cryo-EM PDB通常不含氢,且残基命名可能不规范:
| 问题 | 解决方案 |
|---|---|
| HIS未指定HID/HIE | 根据氢键环境手动重命名,或用 propKa 预测(pH 7.4) |
| ASP/GLU质子化不明 | 默认去质子化(-1),除非位于疏水口袋(如酶活性位点) |
| 非标准残基(MSE, HYP) | 在tleap中映射为标准残基(如MSE→MET) |
| 缺失氢原子 | 由tleap自动添加,切勿用外部工具加氢(可能破坏几何) |
3. 清洗与格式标准化
使用 pdb4amber(AmberTools)进行预处理:
pdb4amber -i cryoem.pdb -o cleaned.pdb \
--reduce \ # 修复常见命名错误
--most-populous \ # 保留最占据的altloc构象
--keep-heterogens # 保留配体/离子此步骤会:
- 删除结晶水、去垢剂(如DUM、OGN);
- 合并多构象为单一构象;
- 标准化残基/原子命名。
四、MD模拟的特殊注意事项
1. 渐进升温平衡
Cryo-EM结构在~100 K下冻结,直接升温至300 K易导致结构崩塌。建议采用分阶段升温+主链约束:
! mdin (Amber)
&cntrl
ntt=3, gamma_ln=2.0,
tempi=100.0, temp0=300.0,
ntb=1, ntp=0, ! NVT系综
ntr=1, restraint_wt=5.0,
restraintmask='@CA,C,N', ! 主链约束
nmropt=1,
/
&wt type='TEMP0', istep1=0, istep2=100000, value1=100.0, value2=300.0 /
&wt type='END' /2. 局部约束策略
根据局部分辨率设置不同强度的约束:
- 高分辨区(<3.5 Å):强约束(5--10 kcal/mol/Ų);
- 中分辨区(3.5--5 Å):弱约束(1--2 kcal/mol/Ų)或仅约束Cα;
- 低分辨区(>5 Å):无约束或删除。
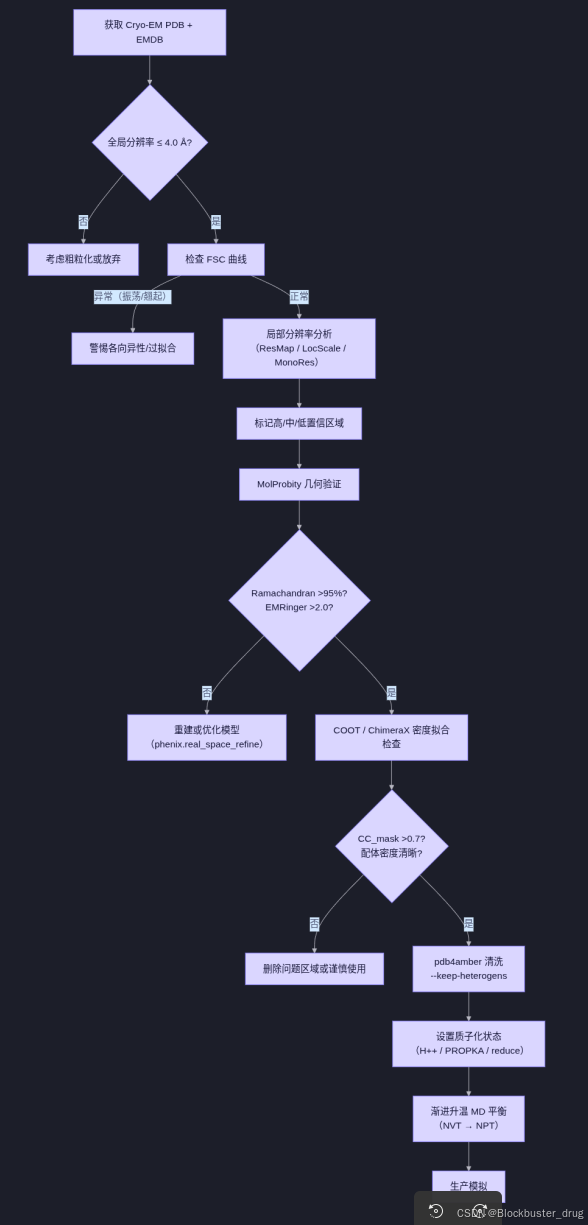
五、实用评估流程图
六、快速检查清单
✅ 通过标准:
- 全局分辨率 ≤ 3.5 Å(功能研究建议 ≤ 3.0 Å);
- 活性位点局部分辨率 ≤ 3.5 Å;
- COOT中模型与密度高度吻合;
- MolProbity:Ramachandran favored >95%,Clashscore <10;
- HIS/ASP/GLU质子化状态合理;
- 使用pdb4amber清洗后交tleap处理。
❌ 危险信号(禁止直接MD):
- 分辨率 >4.5 Å 但建模了精细侧链;
- 配体无清晰密度支持;
- FSC曲线异常或未提供;
- 未提供局部分辨率图;
- Clashscore >20 或 Ramachandran favored <90%。
结语
Cryo-EM为我们打开了观察大型、柔性生物机器的大门,但其结构并非"即插即用"。严谨的质量评估是连接静态结构与动态模拟的桥梁。通过结合定量指标(Ramachandran favored、Clashscore、Rotamer outliers)与人工检查(密度拟合、生物合理性),我们可以识别高置信区域,规避建模陷阱,从而确保MD模拟建立在可靠的物理基础上。
记住:好的模拟始于好的结构。花一小时评估,可节省数百小时无效计算。