目录
[5.1 消息函数](#5.1 消息函数)
[5.2 虚拟图元素](#5.2 虚拟图元素)
[5.3 读出函数](#5.3 读出函数)
[5.4. 多塔结构](#5.4. 多塔结构)
图神经网络概览:图神经网络分享系列-概览
上一篇文章:图神经网络分享系列-MPNN(Neural Message Passing for Quantum Chemistry)(一)
三、相关研究
尽管量子力学在理论上允许计算分子的性质,但物理定律导出的方程过于复杂而无法精确求解。科学家开发了一系列量子力学近似方法,在速度和精度之间权衡,例如采用多种泛函的密度泛函理论(DFT)(Becke, 1993; Hohenberg & Kohn, 1964)、GW近似(Hedin, 1965)和量子蒙特卡洛方法(Ceperley & Alder, 1986)。尽管DFT被广泛使用,但其计算速度对大体系仍显不足(计算复杂度为O(Ne³),Ne为电子数),且相对于薛定谔方程的精确解存在系统性和随机误差。例如,在单核Xeon E5-2660(2.2 GHz)上运行Gaussian G09(ES64L-G09RevD.01)计算一个含9个重原子的QM9分子需约1小时,而17个重原子分子则需8小时(Bing et al., 2017)。
经验势能方法(如Stillinger-Weber势,1985)虽快速且精确,但需针对每种新原子组合从头构建。Hu等(2003)利用神经网络近似DFT中复杂的交换关联势以提高精度,但未改进计算效率,且依赖大量人工设计的原子描述符。
Behler & Parrinello(2007)和Rupp等(2012)提出绕过DFT直接近似量子力学解的方法。前者使用单隐层神经网络预测硅熔体的能量和力以加速分子动力学模拟;后者采用核岭回归(KRR)预测广泛分子的原子化能,两者均依赖手工特征(对称函数和库仑矩阵)以嵌入物理对称性。后续研究用神经网络替代了KRR。
这些方法的特征设计存在固有局限:Behler & Parrinello的表示虽对图同构不变,但难以处理超过三种原子的体系且无法泛化至新组合;Rupp等的表示则需通过数据增强让模型学习同构不变性。
除第2节讨论的八种MPNN外,其他图数据机器学习方法也利用对称性。例如Niepert等(2016)和Rupp等(2012)通过预处理构建标准图表示再输入分类器;Scarselli等(2009)提出在图上运行至收敛的消息传递机制,而非MPNN的固定步长。
四、QM9数据集
为研究MPNN(消息传递神经网络)在预测化学性质上的表现,采用公开的QM9数据集(Ramakrishnan等,2014)。该数据集中的分子由氢(H)、碳(C)、氧(O)、氮(N)和氟(F)原子组成,最多包含9个重原子(非氢原子),总计约13.4万个类药物有机分子,覆盖广泛的化学多样性。每个分子通过密度泛函理论(DFT)计算获得合理的低能结构,因此包含原子空间位置信息,同时计算了多种基础且重要的化学性质。鉴于QM9中部分性质的 foundational 特性,若无法对其性质实现准确统计预测,则更难应对更具挑战性的化学任务。
性质分类
预测的性质可分为四大类:
1. 原子结合能相关性质
反映分子内原子结合的紧密程度,表征不同温度与压力下分解分子所需的能量,包括:
- 0K下的原子化能 ( U_0 )(单位:eV)
- 室温下的原子化能 ( U )(eV)
- 室温下的原子化焓 ( H )(eV)
- 原子化自由能 ( G )(eV)
2. 分子振动相关性质
涉及分子基础振动模式,包括:
- 最高基频振动频率 (
)(单位:
- 零点振动能(ZPVE)(单位:eV)
3. 电子状态相关性质
描述分子中电子的能量状态,包括:
- 最高占据分子轨道(HOMO)能量 (
- 最低未占分子轨道(LUMO)能量 (
- 电子能隙 (
4. 电子空间分布相关性质
量化电子在分子中的空间分布特性,包括:
- 电子空间范围 (
- 偶极矩范数 (
- 静态极化率范数 (
更详细的属性说明可参考补充材料。
五、MPNN变体
探索MPNN时以GG-NN模型为基线,重点关注消息函数、输出函数的设计、输入表示的优化及超参数调谐。后文用d 表示图中每个节点的隐藏表示维度,n表示节点数量。
实现中,MPNN默认处理带独立输入/输出边通道的有向图,消息m 为输入消息m_in 和输出消息m_out 的拼接(参考Li et al., 2016)。应用于无向化学图时,将每条原始边视为同标签的输入与输出双向边。需注意边的方向仅影响参数共享,无向图的消息通道尺寸因此扩展为2d 而非d。
模型输入包括节点特征向量x_v 和带向量值的邻接矩阵A (编码分子键类型及原子间空间距离)。实验也测试了GG-NN族的离散边标签消息函数,此时A 的条目属于大小为k 的离散字母表。初始隐藏状态h_v^0 设为原子特征向量x_v 并填充至维度d 。所有实验均采用时间步t的权重共享,并使用GG-NN族的GRU(Cho et al., 2014)作为更新函数。
5.1 消息函数
矩阵乘法 :初始采用GG-NN的消息函数,定义为方程 ( )。
边网络 :为支持向量值边特征,提出消息函数 ( ),其中 (
) 是将边向量 (
) 映射到 (
) 矩阵的神经网络。
配对消息 :矩阵乘法规则的特点是,节点 ( w ) 到节点 ( v ) 的消息仅依赖于隐藏状态 ( ) 和边 (
),与目标节点状态 (
) 无关。理论上,若允许消息同时依赖源节点和目标节点,可提升消息通道效率。因此尝试了变体消息函数 (
),其中 ( f ) 为神经网络。
在有向图 中,上述消息函数分为 ( ) 和 (
),具体应用取决于边的方向。
5.2 虚拟图元素
探索了两种改进消息传递的方法:
虚拟边类型 :为未连接的节点对添加独立的虚拟边类型,通过数据预处理实现,允许信息在传播阶段远距离传递。
主节点 :引入潜在"主节点",通过特殊边类型与图中所有输入节点连接。主节点作为全局共享空间,每一步消息传递中节点均可读写。主节点可拥有独立维度 ( ) 及内部更新函数(如GRU)的独立权重。该方法复杂度为 (
),理论上能提升模型容量而不显著影响性能。
5.3 读出函数
测试了两种读出函数:
GG-NN读出 :基于方程4定义。
Set2Set模型 :专为集合操作设计,比简单求和节点状态更具表达能力。该模型先对元组 ( ) 做线性投影,输入投影后的集合 ( T ),经 ( M ) 步计算后生成与元组顺序无关的图级嵌入 (
),最终通过神经网络输出结果。
5.4. 多塔结构
MPNN存在可扩展性问题。对于稠密图,单步消息传递阶段需要进行次浮点乘法运算。当节点数
或嵌入维度
增大时,计算开销会显著增加。
为解决该问题,将d维节点嵌入拆分为k组d/k维嵌入
,每组独立运行传播步骤,生成临时嵌入
,并为每组使用独立的消息函数和更新函数。最终通过以下方程将k组临时嵌入混合:
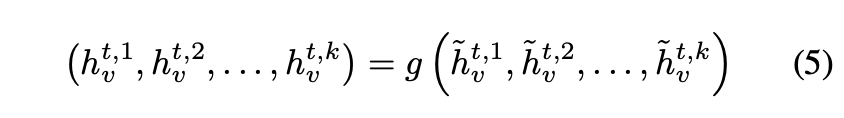
g表示一个神经网络,(x, y, ...)表示拼接操作,且g在图中的所有节点之间共享。这种混合方式保持了节点排列的不变性,同时允许图的不同副本在传播阶段相互通信。其优势在于能够以相同参数量实现更大的隐藏状态,从而在实际中提升计算速度。
例如,当消息函数为矩阵乘法时(如GG-NN),单个副本的传播步骤时间复杂度为O(n²(d/k)²),而共有k个副本,因此总体时间复杂度为O(n²d²/k),混合网络会引入少量额外开销。当k=8、n=9且d=200时,相比k=1、n=9和d=200的架构,推理速度可提升2倍。这种变体对处理更大分子(如GDB-17数据库中的分子)尤为有效。
六、输入表示

分子中每个原子具有多种特征,涵盖原子内电子属性及其参与的化学键信息。完整特征列表参见表1。实验中将氢原子显式作为图中的节点(而非仅将数量作为节点特征),此时图的节点数最多可达29个。需注意,更大的图会显著增加训练时间(约10倍)。邻接矩阵根据模型不同采用三种边表示方式:
化学图:无距离信息时,邻接矩阵条目为离散键类型(单键、双键、三键或芳香键)。
距离分箱:矩阵乘法消息函数需离散边类型,因此将键距离划分为10个区间:在2, 6区间内均匀划分8个箱,额外添加0, 2和6, ∞两个箱。分箱依据所有距离的直方图手动确定。邻接矩阵条目为14种符号,表示键合原子的键类型或非键合原子的距离分箱。实验发现键合原子的距离几乎完全由键类型决定。
原始距离特征:若消息函数处理向量值边,邻接矩阵条目为5维向量:第一维表示原子对的欧氏距离,其余四维为键类型的独热编码。
七、训练方法
每个模型与目标组合通过50次均匀随机超参数搜索进行训练。参数T范围限制为3 ≤ T ≤ 8(实际应用中T ≥ 3均可)。set2set计算次数M选自1 ≤ M ≤ 12。所有模型使用SGD与ADAM优化器训练,批量大小为20,共300万步(约540轮)。初始学习率在1e−5至5e−4间均匀选择,并采用线性衰减:衰减起点为训练进度的10%至90%,衰减因子F ∈ 0.01, 1,最终学习率为初始值l乘以F。
QM-9数据集含130462个分子,随机选取10000个样本作为验证集,10000个作为测试集,其余用于训练。验证集用于早停和模型选择,测试集用于评估。所有目标值归一化为均值0、方差1。训练时最小化模型输出与目标的均方误差,但评估时采用平均绝对误差。
八、实验结果
在所有表格中,报告的是模型平均绝对误差(MAE)与各目标化学精度估计值的比值。若误差比值小于1,则表明模型对该目标达到了化学精度。补充材料中列出了各目标的化学精度估计值,这些数值与Faber等人(2017)的研究一致。模型的MAE可通过公式计算:
(误差比值) × (化学精度)
除非特别说明,所有表格展示的是针对单个目标独立训练的模型结果(而非联合训练13个目标的单一模型)。
模型优化与输入表征
通过大量实验确定了最佳MPNN架构和输入表征方式。实验发现:
- 包含完整的边特征向量(键类型、空间距离)并将氢原子显式作为图节点,对多个目标至关重要。
- 针对单个目标独立训练的模型性能始终优于联合训练13个目标的模型,部分情况下提升幅度达40%。
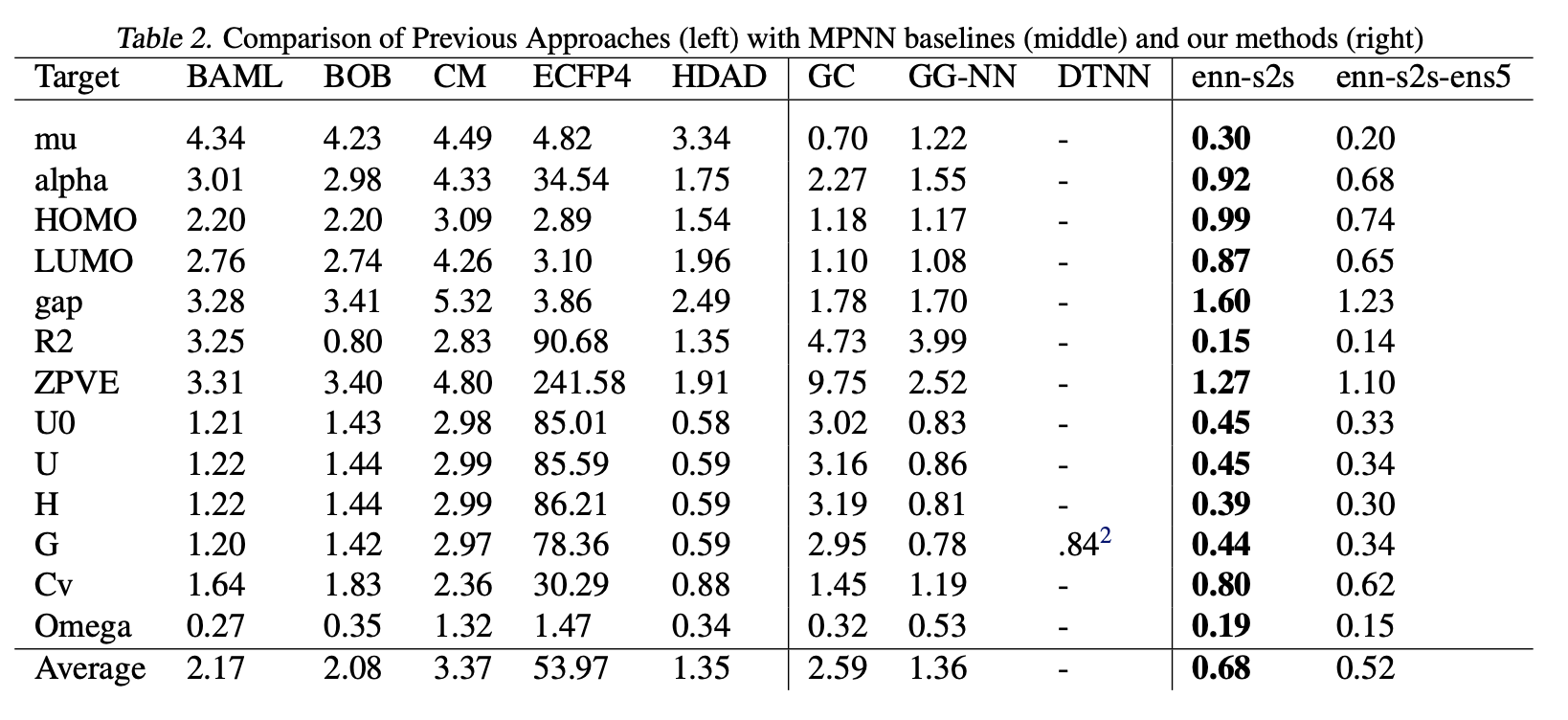
最佳MPNN变体采用边网络消息函数、Set2Set输出,并在显式氢原子图上运行。通过集成验证误差最低的五个模型,测试集性能得到进一步提升。
与现有方法的对比
表2比较了最佳MPNN变体(enn-s2s)及其集成模型(enn-s2s-ens5)与Faber等人(2017)报告的先前最优结果。非集成模型的最佳误差比值以粗体显示。基线方法包括五种手工设计的分子表征(如库仑矩阵、键袋、扩展连通性指纹等)及两种现有MPNN模型(GC和GG-NN)。新MPNN在13个目标中的11个上达到化学精度,并在全部目标上实现最优性能。
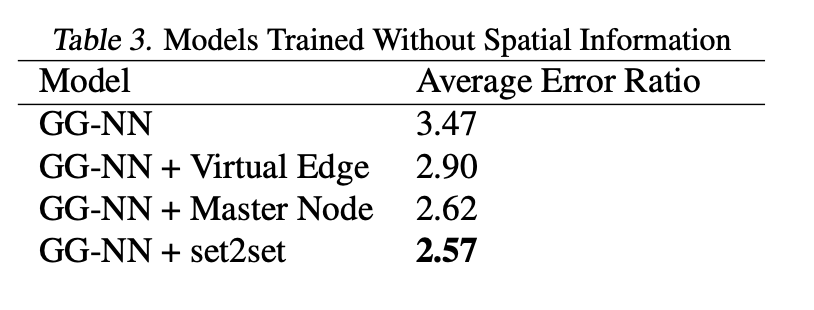
无空间信息的训练
实验验证了输入中不含空间信息时的表现。结果表明,通过增强MPNN捕获图中节点间长程相互作用的能力可显著提升性能。四种实验设置包括:稀疏图训练的GG-NN、添加虚拟边、添加主节点、改用Set2Set输出。Set2Set输出在13个目标中的5个上达到化学精度(表3)。
多塔结构(Towers)
开发多塔变体的初衷是缩短训练时间并支持更大图的训练,但其亦表现出泛化性能的提升。表4显示,在联合训练和单目标训练中,采用多塔结构的GG-NN + Set2Set输出模型在13个目标中的12个上优于基线模型。多塔结构的优势可能类似于模型集成,但将其与边网络消息函数结合的尝试未能进一步改进性能。
注:表3和表4的实验使用了DFT计算输出的部分电荷特征(实际应用中不可用),而表2的最优结果未使用此特征。更多细节见补充材料。
额外实验
在初步实验中,尝试解耦不同时间步的权重共享,但发现提升性能最有效的方式仍是保持权重共享并增大隐藏维度 ( d )。早期实验还表明,基于配对的消息函数(pair message function)性能逊于边网络函数(edge network function),甚至在专为配对消息设计的路径查找玩具问题上也表现不佳。在联合训练13个目标任务时,边网络函数在11项任务中优于配对消息,其平均错误率比为1.53,而配对消息为3.98。由于训练难度较高,后续未继续研究该函数。小规模训练集的性能数据可参考补充材料。
九、结论与未来工作
结果表明,配备合适消息、更新和输出函数的MPNN(消息传递神经网络)在预测分子性质时具有有益的归纳偏差,其性能超越多个强基线模型,且无需复杂特征工程。研究还揭示了允许图中节点通过主节点(master node)或set2set输出进行长程交互的重要性。塔式结构(towers variation)提升了模型可扩展性,但需进一步改进以应对更大规模图结构。
未来重要方向包括设计能泛化至训练集外更大规模图的MPNN,或至少构建能暴露跨图规模泛化问题的基准测试。使用空间信息时,泛化至更大分子结构尤为困难:其一,原子间距离分布高度依赖原子数量;其二,当前最成功的空间信息使用方法会生成全连接图,导致输入消息数量与节点数相关。针对后者,探索在输入消息向量上引入注意力机制可能是潜在解决方案。
好了,本篇论文已经基本完成讲解,下篇文章会描述附录部分~