文献介绍

文献题目: 从空间转录组数据估计肿瘤中的细胞谱系
研究团队: Peng Jiang(美国国家癌症研究所)
发表时间: 2023-02-02
发表期刊: Nature Communications
影响因子: 16.6(2023年)
DOI: 10.1038/s41467-023-36062-6
摘要
空间转录组学(ST)技术通过原位捕获实现了肿瘤组织的拓扑基因表达谱分析。然而,每个捕获点可能包含多种免疫细胞和恶性肿瘤细胞,且不同组织区域的细胞密度存在差异。针对现有方法在解析通用 ST 或大块肿瘤数据时面临的挑战,肿瘤 ST 数据中的细胞类型反卷积仍具难度。作者开发了肿瘤空间细胞估计器(SpaCET),用于从肿瘤 ST 数据中推断细胞身份。SpaCET 首先通过整合常见恶性肿瘤中拷贝数变异和表达变化的基因模式字典来评估癌细胞丰度,随后通过约束回归模型校准局部细胞密度并确定免疫细胞(immune)与基质(stromal)细胞谱系占比。基于模拟数据及匹配双盲组织病理学标注作为金标准的真实 ST 数据验证,SpaCET 的准确性显著优于现有方法。进一步将细胞分数与配体-受体共表达分析相结合,SpaCET 揭示了肿瘤-免疫界面(tumor-immune interface)的细胞间相互作用如何促进癌症进展。
前言
在空间背景下分析细胞转录组,对于理解肿瘤进展和治疗耐药性的机制至关重要。近年来,空间转录组学(ST)发展迅速,其基因覆盖范围从数个靶标扩展至全基因组,分辨率也从亚细胞水平到多细胞水平各异。作为 ST 方法的一个重要分支,基于位置分子条形码的原位捕获策略能够无偏地捕获完整组织中的全转录组。其代表性技术包括 Slide-seq、10x Visium,以及 Visium 所基于的早期原位捕获方法。具体而言,商业化的 Visium 平台可以分析新鲜冷冻组织和福尔马林固定石蜡包埋(FFPE)组织中的 mRNA 水平,使其得到广泛应用。然而,各种捕获策略的空间点(直径为 10--100 微米)可能会测量来自不同谱系多个细胞的混合信号。因此,解析点内的细胞身份是刻画组织空间细胞图谱的关键步骤。
现有许多方法可用于解析一般 ST 数据和整体转录组图谱中的细胞类型。然而,这些方法及其底层策略在处理肿瘤 ST 数据的独特问题时面临挑战。一些方法,如 Stereoscope、RCTD 和 CIBERSORTx,依赖合适的恶性细胞参考图谱来预测癌细胞比例。其他方法,如 EPIC,通过估算未被预定义细胞特征覆盖的未知细胞比例来估算无参考的恶性细胞比例。然而,该策略无法区分恶性细胞与真正的未知细胞类型。此外,不同肿瘤区域的细胞密度可能存在显著差异;因此,现有方法通过将每个点内的总比例归一化为 1 所解析出的细胞比例,在不同位置之间不具有可比性。
另一种反卷积细胞比例的策略是生成与同一肿瘤样本 ST 数据配对的单细胞 RNA 测序(scRNA-seq)数据。然而,在冷冻或 FFPE 组织样本中进行单细胞实验具有挑战性,因为这需要新鲜样本并增加额外成本。即使有新鲜样本,scRNA-seq 也可能无法可靠地捕获某些细胞类型,例如中性粒细胞,因为它们对快速的 RNA 损伤非常敏感。 本研究介绍了一个计算框架 SpaCET(Spatial Cellular Estimator for Tumors) ,用于解析肿瘤 ST 数据中的细胞身份。SpaCET 应对了肿瘤异质性、组织密度变化、免疫细胞完整性以及亚谱系间共线性等挑战,而现有反卷积方法对这些因素的考虑尚不充分。基于双盲组织病理学注释,SpaCET 在涵盖七种癌症类型的八个肿瘤 ST 数据集上均表现出优于其他方法的性能。此外,SpaCET 揭示了若干支持肿瘤进展的潜在细胞-细胞相互作用。SpaCET 的源代码公开于 https://github.com/data2intelligence/SpaCET。
研究结果
1. 肿瘤空间转录组学中分解细胞谱系
SpaCET 框架通过三个阶段估算肿瘤空间转录组数据中的细胞谱系和细胞间相互作用(Fig. 1a)。
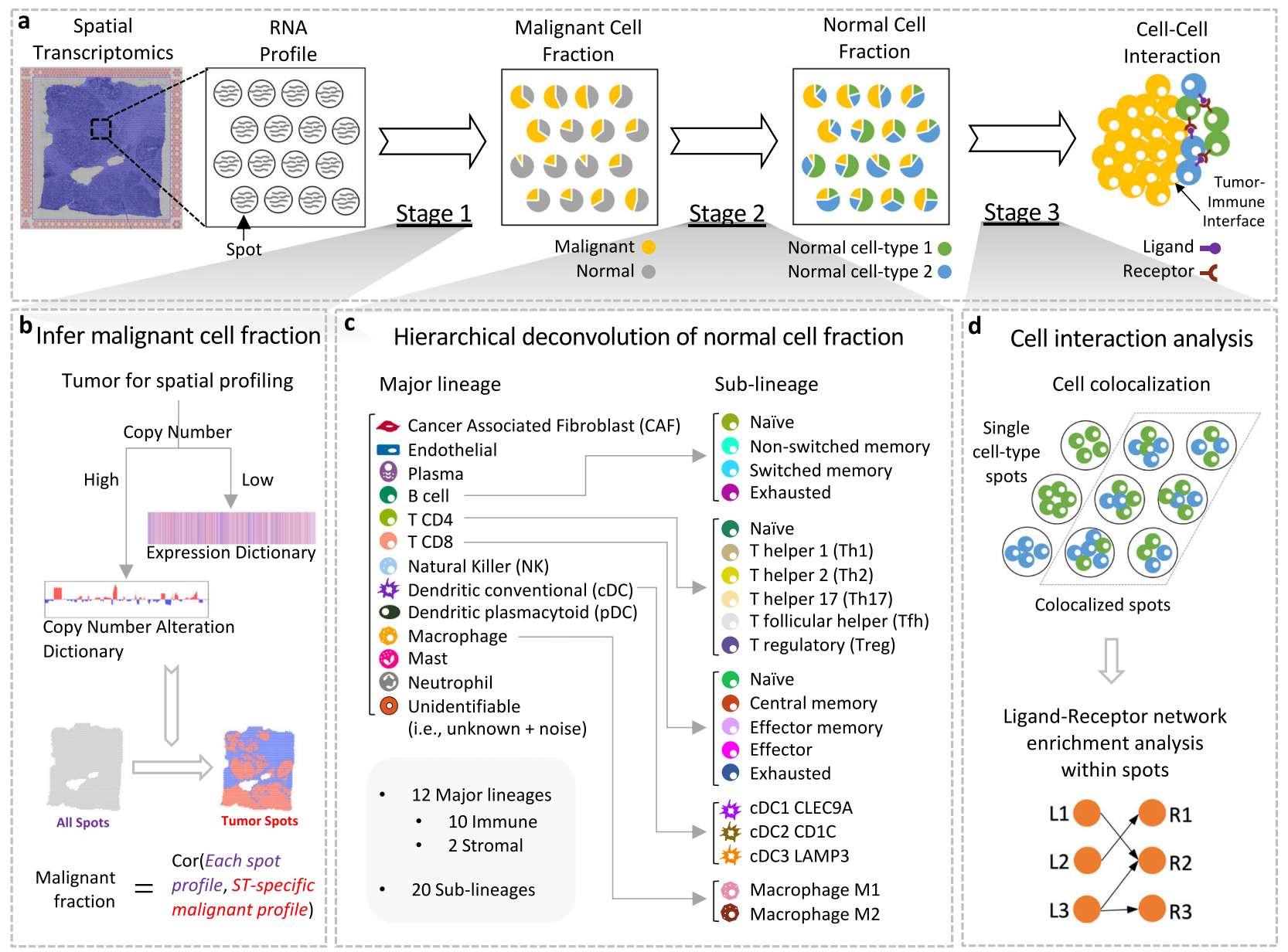 Figure 1. 推断肿瘤空间转录组学中的细胞组分和相互作用
Figure 1. 推断肿瘤空间转录组学中的细胞组分和相互作用
(A) 从输入空间转录组学(ST)数据到细胞谱系比例和细胞间相互作用的三个阶段。
(B) 通过基因模式字典推断恶性细胞比例。对于肿瘤 ST 数据集,SpaCET 使用拷贝数变异或肿瘤转录组模式的字典来识别肿瘤点,并进一步计算 ST 特异性的恶性表达谱。然后,SpaCET 将 ST 特异性的恶性表达谱与每个点的表达谱进行关联,并将相关系数归一化到 0--1,作为所有点的恶性比例。
(C) 非恶性细胞比例的层次反卷积。基于来自公共 scRNA-seq 数据图谱的分层细胞参考,SpaCET 利用约束线性回归在两个层次上估算细胞比例。在第一层,SpaCET 将非恶性细胞比例分解为主要谱系和不可识别组分。在第二层,主要谱系比例被进一步分解为相应的亚谱系比例。
(D) 通过测试细胞共定位和配体-受体相互作用进行细胞间相互作用分析。基于推断出的细胞比例,SpaCET 通过点间的相关性来测量细胞共定位。然后,对于细胞类型共定位的点,SpaCET 检验配体-受体共表达的显著性,作为物理相互作用的进一步证据。
首先,SpaCET 基于一个基因模式字典来估算恶性细胞比例,该字典涵盖了常见肿瘤类型中的拷贝数变异(CNA)和恶性转录组特征 (Fig. 1b and Supplementary Fig. 1a)。大多数肿瘤 ST 数据集没有匹配的 scRNA-seq 数据作为恶性细胞参考。然而,多数人类肿瘤的一个一致特征是染色体不稳定性,这导致每种癌症中都有常见的 CNA 模式。此外,在 CNA 水平较低的染色体稳定型肿瘤中,恶性细胞可能仍具有区分肿瘤与正常细胞的转录组特征。因此,作者利用来自癌症基因组图谱(TCGA)中涵盖 30 种肿瘤类型的大约 10,000 个患者样本,创建了一个 CNA 或肿瘤-正常表达差异的基因模式字典(Supplementary Fig. 1b, c and Supplementary Table 1)。在每个肿瘤 ST 数据中,SpaCET 会搜索其表达谱与相关肿瘤类型的 CAN 或表达模式相关的恶性细胞点。
其次,SpaCET 在一个统一的线性模型下反卷积非恶性细胞比例并调整细胞密度 (Fig. 1c)。利用来自不同癌症类型的 scRNA-seq 数据集,作者定义了分层谱系中免疫细胞和基质细胞的参考表达谱(Fig. 1c and Supplementary Table 2)。SpaCET 利用约束线性回归在两个层次上估算细胞谱系。SpaCET 首先将非恶性细胞比例分解为免疫谱系(immune)、基质谱系(stromal)以及细胞谱系参考无法解释的"不可识别(unidentifiable)"组分(Fig. 1c)。"不可识别"类别使线性模型能够减少对细胞含量低或存在未知细胞类型的 ST 点的估算细胞比例。然后,SpaCET 在受其父系谱系比例约束的条件下,进一步反卷积免疫亚谱系的比例(Fig .1c)。密切相关细胞类型的表达特征可能导致共线性,从而引起结果的高度变异。分层分解方案将限制因共线性导致的亚谱系结果变异,使其不影响更高层次的细胞比例。
第三,SpaCET 基于细胞共定位和配体-受体共表达分析推断细胞间相互作用 (Fig. 1d)。作者关注同一 ST 点内细胞之间的紧密接触,而非不同 ST 点之间的接触,因为点间的间隙(例如,Visium 平台约为 50 微米)可能跨越多个细胞。作者计算所有 ST 点上细胞比例的线性相关性,以评估细胞类型的共定位。高度正相关表明细胞类型对倾向于共定位在一起。为了推断物理相互作用,作者进一步针对共定位的细胞类型对,测试配体与受体基因在同一 ST 点内的共表达情况。
2. 通过模拟 ST 数据进行性能验证
为了评估 SpaCET 的性能,作者对模拟的(本节)和带有双盲组织病理学注释的真实 ST 数据(下一节)进行了反卷积。模拟 ST 数据集通过从一个 scRNA-seq 数据集中混合 3-10 个单细胞转录组图谱来生成,以模拟一个点的信号。因此,每个合成 ST 点中细胞类型的实际比例是已知的。作为评估指标,作者计算了反卷积得到的细胞比例与模拟中实际细胞混合比例之间的皮尔逊相关系数(r)和均方根误差(RMSE)。
作者收集了来自黑色素瘤、乳腺癌、结直肠癌、头颈癌、肝癌以及非小细胞肺癌的 10 个 scRNA-seq 数据集(Supplementary Table 2)。每项研究都包含来自不同细胞谱系的数千个单细胞,这使我们能够构建一个全面的肿瘤微环境细胞图谱(Fig. 2a, b and Supplementary Fig. 2a)。作者进行了数据集内部和数据集之间的验证,以评估对合成 ST 数据的反卷积性能。对于数据集内部验证,每个 scRNA-seq 数据集按患者均分为两组。一组患者用于生成细胞参考图谱,另一组不重叠的患者用于生成合成 ST 数据。对于数据集之间验证,参考图谱和合成 ST 数据则来自不同的 scRNA-seq 数据集。
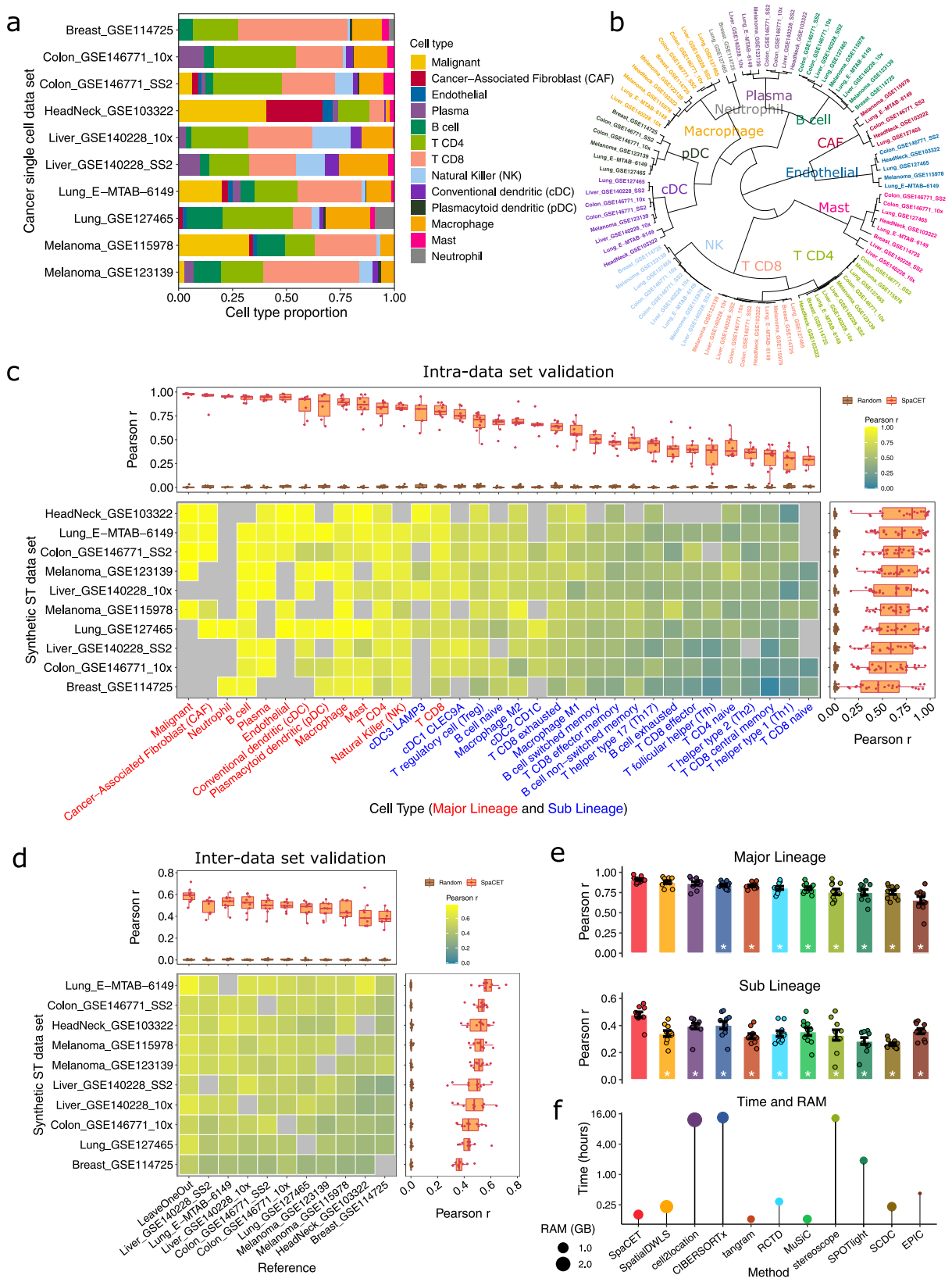 Figure 2. 基于模拟 ST 数据的性能评估
Figure 2. 基于模拟 ST 数据的性能评估
(A) 用于 ST 模拟的 10 个肿瘤 scRNA-seq 数据集中的细胞谱系比例。
(B) 基于标记基因集相似性,对来自 scRNA-seq 数据集的细胞谱系参考图谱进行层次聚类。
(C) 每个 scRNA-seq 数据集(行)和细胞类型(列)在数据集内部验证中的性能。热图中的颜色表示预测细胞比例与已知细胞比例之间的皮尔逊相关系数(r)。热图中的灰色表示该 scRNA-seq 数据集中缺失的细胞类型。顶部的箱线图展示了同一细胞类型在所有数据集中的 r 值(n=10)。右侧的箱线图展示了同一数据集中所有细胞类型预测的 r 值。对每个合成 ST 数据内的细胞类型比例向量的点身份进行了随机打乱,并计算 r 值作为随机对照。
(D) scRNA-seq 队列之间的数据集间验证性能。列标签和行标签分别显示用于生成细胞类型参考图谱和合成 ST 数据的 scRNA-seq 数据集(n=10)。热图中的颜色表示所有细胞类型的预测比例与已知比例之间的中位数皮尔逊相关系数(r)。箱线图和随机对照的绘制方式同图 C。
(E) SpaCET 与先前方法的性能比较(颜色顺序与图 F 一致)。一个点代表从单个 scRNA-seq 数据集合成的模拟 ST 数据集(n=10)。一个 ST 数据集的 y 值表示所有细胞类型的预测比例与已知比例之间的中位数皮尔逊相关系数 r。所有工具均使用图 D 中的留一法特征。通过双侧 Wilcoxon 符号秩检验评估 SpaCET 与其他工具之间的差异。星号表示 SpaCET 显著优于其他方法(BH 校正后 p 值 <0.05)。柱高表示模拟 ST 数据集的平均值;误差线表示标准误。
(F) 使用默认参数、包含 1200 个点的模拟 ST 数据集,比较运行时间和内存消耗。
大多数数据集内部的细胞类型反卷积验证都达到了高准确度(Fig. 2c and Supplementary Fig. 2b,右侧箱线图)。如果模拟 ST 数据的 read counts 保持在单细胞参考数据的 50% 范围内,其性能也表现稳健(Supplementary Fig. 2c)。此外,特定谱系,如恶性细胞(malignant cells)、癌症相关成纤维细胞(CAFs)、中性粒细胞(neutrophil cells)、B细胞、浆细胞(plasma cells),具有较高的预测准确度(Fig. 2c,上方箱线图)。然而,密切相关的细胞类型反卷积具有挑战性,因为亚谱系的性能指标低于其父系谱系(Fig. 2c)。
在数据集之间的验证中,在一个单细胞队列中生成的参考图谱通常可以预测由其他单细胞队列合成的 ST 数据中的细胞比例(Fig. 2d and Supplementary Fig. 2d,上方箱线图)。作者还生成了留一法特征用于数据集之间的评估,即使用除被留作合成测试 ST 数据之外的所有数据集来创建参考图谱。平均而言,留一法特征的表现优于来自单个 scRNA-seq 数据集的个体图谱,这表明整合多个 scRNA-seq 数据集可以为多种肿瘤类型的细胞反卷积创建一个具有泛化能力的参考(Fig. 2d)。此外,采用分层回归的 SpaCET 优于在单一反卷积水平上分解所有亚谱系的方法(Supplementary Fig. 2e)。因此,在后续分析中,作者使用了一个组合的分层参考,即对来自 10 个 scRNA-seq 数据集的每种细胞类型的转录组图谱进行平均。
作者还将 SpaCET 与几种具有代表性的 ST 和整体细胞类型解卷积方法在分解合成 ST 数据方面进行了比较。在模拟环境中,SpaCET 在主要谱系和亚谱系上的表现均优于其他方法(Fig. 2e and Supplementary Fig. 3)。此外,作者计算了所有方法的运行时间和内存消耗,SpaCET 属于高效算法组之一(Fig. 2f)。
3. 通过真实 ST 数据和双盲组织病理学注释进行验证
接下来,作者使用真实的肿瘤 ST 数据评估了 SpaCET 的性能。生成 ST 图谱通常会提供来自同一组织切片的苏木精-伊红(H&E)染色图像。根据 H&E 形态学特征,病理学家在不了解任何反卷积结果的情况下,标记了局部组织密度以及肿瘤区、间质区、淋巴细胞区和巨噬细胞区。作者将 SpaCET 应用于分解涵盖七种肿瘤类型的八个肿瘤 ST 数据集(Fig. 3a and Supplementary Table 3)。双盲病理学注释与估算细胞比例之间的一致性将表明细胞类型解卷积的准确性。
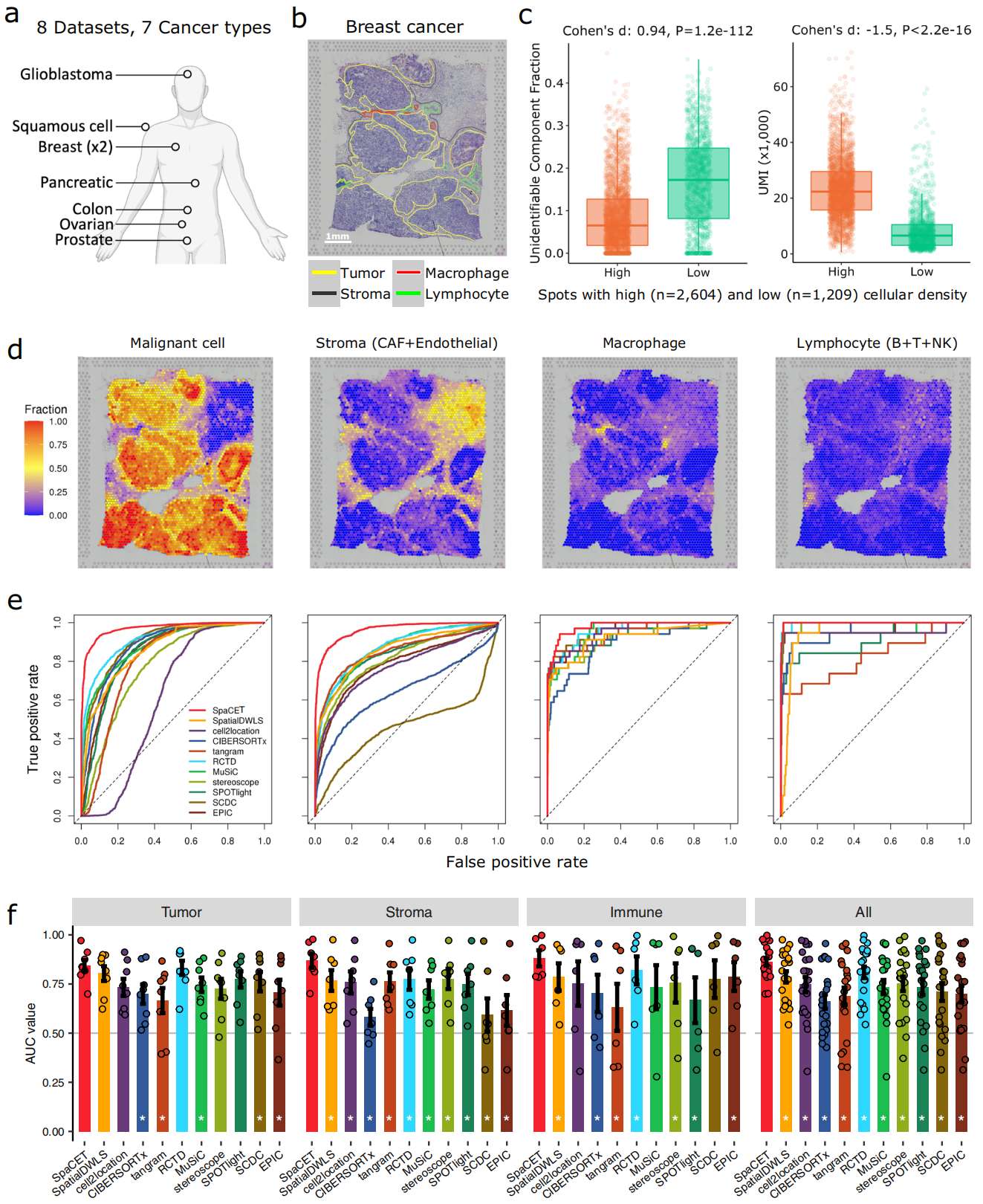 Figure 3. 基于双盲病理注释的性能验证
Figure 3. 基于双盲病理注释的性能验证
(A) 用于性能评估的多个肿瘤 ST 数据集。人体轮廓图使用 BioRender 生成。
(B) 示例苏木精-伊红(H&E)图像及双盲病理学注释。
(C) 高细胞密度区和低细胞密度区中,各点上的不可识别组分比例(左图)和唯一分子标识符(UMI)计数(右图)。通过计算 Cohen's d 效应量和双侧 Wilcoxon 秩和检验来比较组间值。
(D) SpaCET 反卷积得到的恶性细胞、基质细胞、巨噬细胞和淋巴细胞的比例。
(E) 细胞比例预测的受试者工作特征(ROC)曲线。该示例基于图 b 中的细胞区域注释。对于每种方法,ROC 曲线展示了在不同点内细胞比例阈值下,假阳性率与真阳性率的关系。
(F) 各方法之间的性能比较。每个点代表一个数据集(每个条形对应n=8)。Y 轴表示每种方法细胞比例解卷积的 ROC 曲线下面积(AUC)值。子图展示了不同肿瘤区域的结果,最后一个子图同时考虑了所有三种区域类型的数据。在每个子图中,通过双侧 Wilcoxon 符号秩检验评估 SpaCET 与其他工具之间的差异。星号表示 SpaCET 显著优于其他方法(BH 校正后 p 值 <0.05)。柱高表示各 ST 数据集的平均值;误差线表示标准误。
例如,来自 10x Visium 的一个乳腺癌 ST 数据集在 3183 个点上测量了 22,953 个基因(Supplementary Fig. 4a)。同一 ST 组织切片的 H&E 染色通过细胞形态实现了肿瘤区、淋巴细胞区和间质区的病理学注释,并通过含铁血黄素沉积实现了巨噬细胞的注释(Fig. 3b)。作者观察到,SpaCET 中乳腺癌特异的 CNA 特征被激活以估算恶性细胞比例,表明该乳腺肿瘤具有染色体不稳定性(Supplementary Fig. 4b)。SpaCET 的反卷积结果显示,该乳腺肿瘤主要由恶性细胞(malignant cells)、癌症相关成纤维细胞(CAFs)、内皮细胞(endothelial cells)、巨噬细胞(macrophages)和 CD4+T 细胞组成(Supplementary Fig. 4c)。
作者观察到,低密度区域具有较大的不可识别组分(Fig. 3c and Supplementary Fig. 4c, d)。一种可能的解释是,稀疏的组织区域存在缺失现象,即许多基因在 ST 数据中的 read counts 为零。这种缺失会带来高噪声,从而降低已知细胞类型的回归系数。事实上,ST 点上的缺失率与病理学家注释的组织密度成正比(Fig. 3c and Supplementary Fig. 4e)。
为简化起见,SpaCET 每个肿瘤 ST 数据集仅提供一种恶性细胞类型。然而,不同空间区域的恶性细胞可能呈现由癌症演变及与局部环境相互作用决定的不同细胞状态(Supplementary Fig. 4f--h)。因此,SpaCET 还提供了识别癌细胞亚状态的附加步骤。
基于 H&E 染色注释,作者使用受试者工作特征(ROC)曲线比较了 SpaCET 与先前方法的预测准确性(Fig. 3d, e)。尽管 SpaCET 不需要恶性细胞特征且内置了正常细胞参考,但大多数先前方法需要单细胞参考来进行反卷积。由于大多数肿瘤 ST 数据没有来自同一样本的匹配 scRNA-seq 数据,作者根据他们的 scRNA-seq 数据收集为现有工具构建了一个泛癌单细胞参考。作者发现,在估算恶性细胞、基质细胞和巨噬细胞方面,SpaCET 优于其他方法(Fig. 3e)。在淋巴细胞方面,几种方法(例如 Stereoscope 和 RCTD)也取得了与 SpaCET 相当的高性能。
作者进一步在七种癌症类型的另外七个 ST 数据集上进行了评估(Fig. 3f and Supplementary Figs. 5--11)。总体而言,SpaCET 在跨细胞类型的估算上比其他方法更准确(Fig. 3f and Supplementary Figs. 12)。SpatialDWLS 和 RCTD 在所有细胞类型上也取得了高性能。除了将 SpaCET 与其他方法进行比较外,作者还评估了 SpaCET 算法设计的鲁棒性和有效性。通过将肿瘤 ST 数据从每个点 4000 个基因降采样到 500 个基因,作者发现 SpaCET 在低质量 ST 数据上仍然保持高性能(Supplementary Fig. 13a)。
对于恶性细胞定量(Fig. 1b and Supplementary Fig. 1a),SpaCET 针对多种肿瘤类型准备了 CNA 和肿瘤-正常差异表达的联合模式字典。由于空间转录组图谱与癌症类型特异的 CNA 模式之间存在显著相关性,作者收集的所有肿瘤 ST 数据都使用 CNA 模式进行癌细胞定量(Supplementary Figs. 4--11)。然而,我们仍然使用这些 ST 数据作为替代,来评估为染色体稳定型肿瘤准备的表达特征。这些表达特征取得了可比较的性能(Supplementary Fig. 13b),支持了我们基于表达的程序(该程序将在 CNA 低的肿瘤中发挥作用)的可靠性。SpaCET 回归模型中的不可识别组分仅在反卷积基质细胞方面带来了微小的性能提升(Supplementary Fig. 13c)。
4. SpaCET 可以分解各种分辨率的 ST 数据
不同的 ST 平台具有不同分辨率的空间捕获点,范围从 10 微米到 100 微米不等。因此,作者评估了 SpaCET 是否适用于更广泛的一系列具有更高和更低分辨率的原位捕获数据。在作者收集的数据集中(Supplementary Table 3),八个中有六个来自 10x Visium 平台,其检测点直径为 55 微米。为了在具有更高空间分辨率的数据上进行评估,作者应用 SpaCET 分析了一个结肠癌 Slide-RNA-seq 数据集,该数据集包含 18,288 个直径为 10 微米的微珠,覆盖 16,270 个基因(Supplementary Fig. 10a, b)。病理学家根据细胞形态对匹配的 H&E 图像进行了肿瘤区和间质区的注释(Fig. 4a)。反卷积结果显示,SpaCET 始终优于其他方法(Fig. 4b, c and Supplementary Fig. 10d)。由于微珠直径为 10 微米,覆盖 1-2 个细胞,SpaCET 能够实现单细胞分辨率的细胞类型图谱(Fig. 4d, e and Supplementary Fig. 10e)。
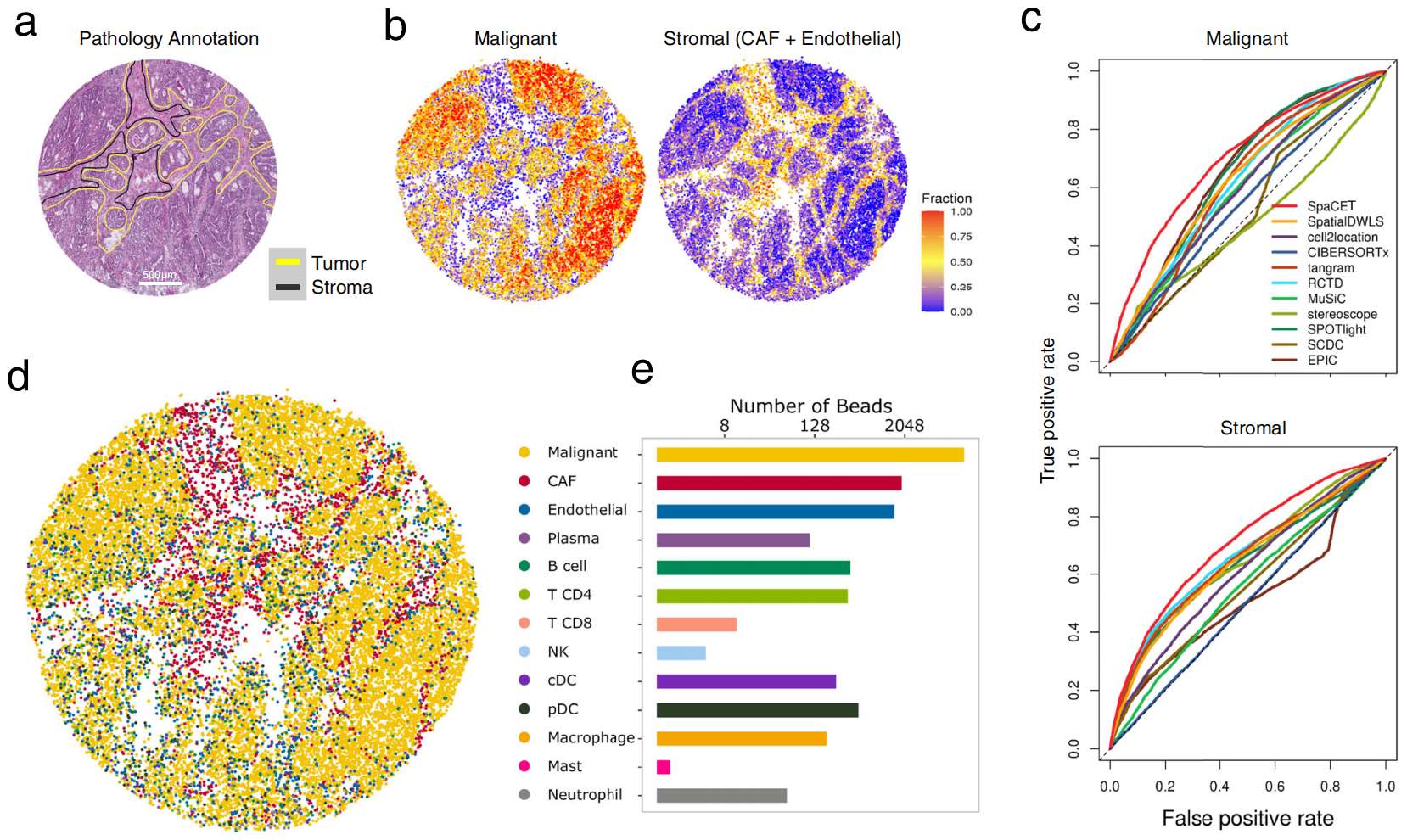 Figure 4. SpaCET 在结肠癌 Slide-seq 数据集上的应用
Figure 4. SpaCET 在结肠癌 Slide-seq 数据集上的应用
(A) 带有双盲病理学注释的 H&E 染色图像。
(B) SpaCET 反卷积得到的恶性细胞和基质细胞比例。
(C) 基于图 a 中注释的细胞比例预测 ROC 曲线,如图 3e 所示。
(D) 主要细胞谱系的空间定位。一个微珠的细胞类型由该微珠中最丰富的细胞类型定义。
(E) 每种细胞类型的微珠数量。
对于低分辨率数据的评估,作者收集了一个通过早期原位捕获方法(Visium 即由此发展而来)生成的胰腺导管腺癌数据集,其点直径为 100 微米(Supplementary Fig. 11)。与其他方法相比,SpaCET 取得了可靠的性能(Supplementary Fig. 12 的最后一行)。根据匹配的 scRNA-seq 数据,该胰腺肿瘤包含腺泡细胞和导管细胞,这些细胞未包含在 SpaCET 的内部细胞类型参考中(Fig. 1c)。SpaCET 可以接受匹配的 scRNA-seq 数据集作为定制的细胞类型参考(Supplementary Fig. 14a)。SpaCET 对腺泡细胞和导管细胞的反卷积结果与其在 H&E 图像中标注的位置高度一致(Supplementary Fig. 14b, c)。
5. SpaCET 揭示空间环境中细胞间的相互作用
肿瘤中的细胞-细胞相互作用在癌症进展和治疗耐药性中发挥着关键作用。单细胞 RNA 测序数据可以通过分析跨细胞类型的配体-受体共表达来揭示细胞间通讯。然而,这种分析丢失了空间邻近信息,因此可能报告在空间中从未接触的假阳性相互作用。SpaCET 的反卷积结果应能在空间背景下实现细胞相互作用分析。
为了从 ST 数据中识别细胞间相互作用,SpaCET 采用了两步法:先评估细胞共定位,再进行配体-受体共表达分析。由于 ST 点之间的间隙可能跨越多个细胞,作者的方法研究了同一 ST 点内的细胞接触,而非不同点之间的接触。作者基于估算的细胞比例,计算了细胞类型对在所有 ST 点上的 Spearman 相关性。细胞类型对呈现强正相关性表明它们存在细胞共定位。例如,在一个乳腺肿瘤中,作者识别出几个潜在的共定位细胞类型对,例如 CAF 与内皮细胞,以及 CAF 与 M2 型巨噬细胞(Fig. 5a and Supplementary Fig. 15a)。
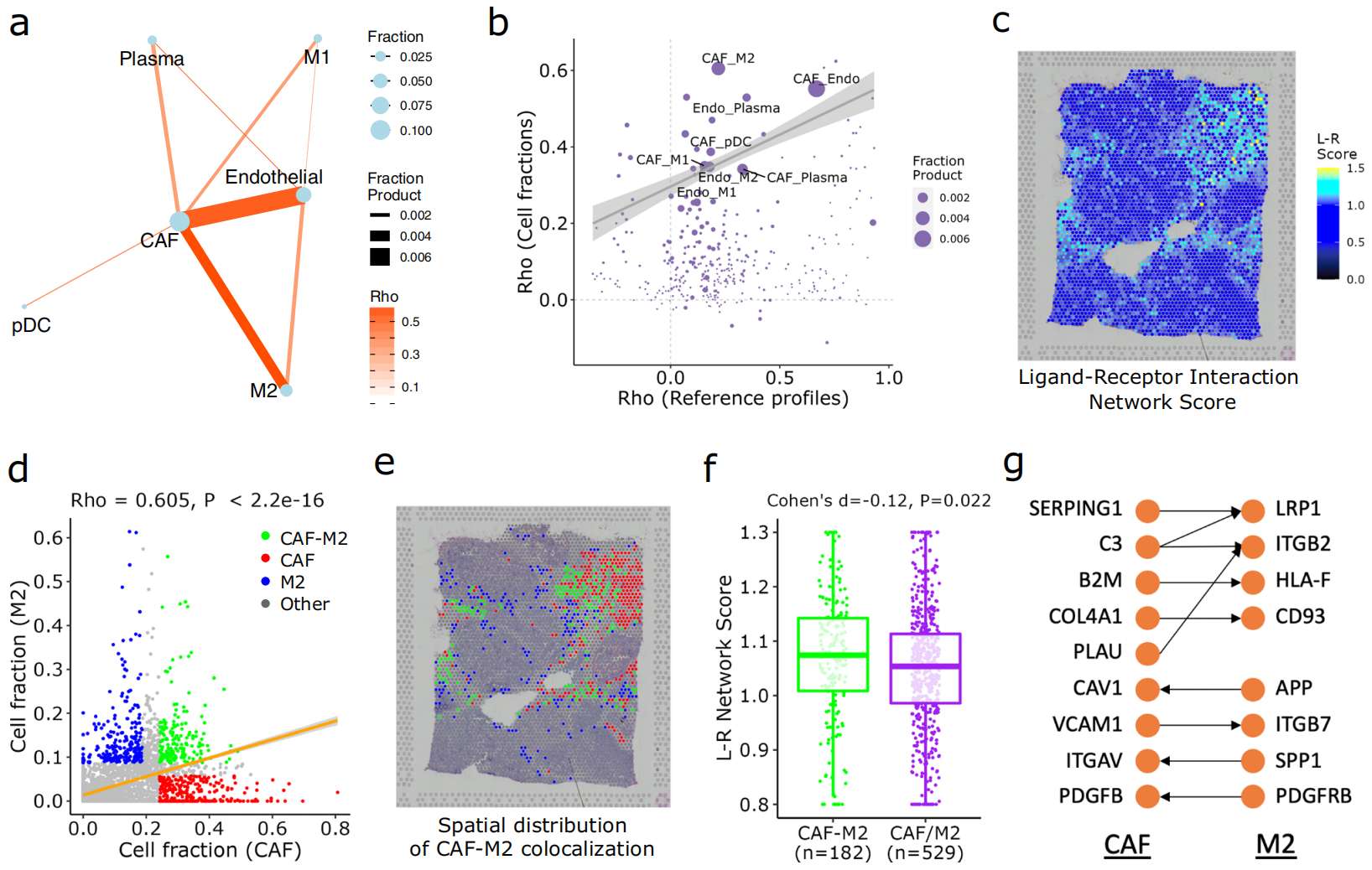 Figure 5. SpaCET 识别乳腺肿瘤中的细胞间相互作用
Figure 5. SpaCET 识别乳腺肿瘤中的细胞间相互作用
(A) 乳腺肿瘤 ST 点中细胞类型比例的 Spearman 相关性。网络中的每个节点代表一种细胞类型,节点大小表示该细胞类型在所有点中的平均比例。每条边代表一个细胞类型对的共定位,边的大小表示该细胞类型对的比例乘积。
(B) 参考图谱之间的 Spearman 相关性(x轴)与细胞类型比例之间的 Spearman 相关性(y轴)的关系。每个点代表一个细胞类型对。直线表示加权线性回归结果,灰色阴影为 95% 置信区间。
(C) 所有点的配体-受体相互作用网络得分。
(D) 所有点中 CAF 和 M2 型巨噬细胞的比例。每个点代表一个 ST 点。根据 CAF 或 M2 细胞比例,将点分为四类:CAF-M2 共定位点(CAF 和 M2 比例均在前 15%,n=182)、CAF 主导点(CAF 比例在前 15% 且 M2 比例在后 75%,n=295)、M2 主导点(M2 比例在前 15% 且 CAF 比例在后 75%,n=234)和其他点(n=3102)。直线表示 CAF 与 M2 细胞比例之间的线性回归,灰色阴影为 95% 置信区间。Rho 值和 p 值通过双侧 Spearman 相关性检验计算(n=3813个点)。
(E) 图 d 中 CAF-M2 共定位点以及 CAF/M2 主导点的空间分布。
(F) 图 d 中 CAF-M2 共定位点与 CAF/M2 主导点之间 L-R 相互作用网络得分的差异。
(G) 在当前乳腺癌组织中介导 CAF-M2 相互作用的 L-R 对。箭头方向从配体指向受体。
为了排除由相似参考图谱导致的高细胞比例相关性,作者比较了细胞类型比例之间的相关性与细胞类型参考图谱之间的相关性。尽管 CAF 和内皮细胞比例之间的相关性较高,但它们的图谱相似性也相应较高(Fig. 5b)。然而,CAF 与 M2 型巨噬细胞参考图谱之间的相似性相对较低(Fig. 5b),这表明 CAF-M2 共定位并非仅仅是由于图谱相似性所致。
6. ST 点内配体-受体相互作用
细胞共定位并不直接指示物理相互作用。因此,作者通过分析 ST 点内的配体-受体(L-R)相互作用,寻求细胞间相互作用的进一步证据。根据先前的一项研究,作者获得了大约 2500 个 L-R 对。作者为每个点计算了一个 L-R 网络得分,该得分是 L-R 基因对之间表达值乘积的总和,并通过具有相同连接度的 1000 个随机 L-R 网络的平均得分进行归一化。
每个 ST 点的 L-R 网络得分指示了该位置配体-受体相互作用的强度(Fig. 5c and Supplementary Fig. 15b),但并不反映细胞类型之间的特异性相互作用。因此,SpaCET 进一步对每种细胞类型对进行了 L-R 网络得分的富集分析。例如,针对乳腺肿瘤组织中 CAF 与 M2 细胞之间的共定位,SpaCET 将所有 ST 点分为四类:CAF-M2 共定位点、CAF 或 M2 主导点,以及其他点(Fig. 5d, e)。作者发现,CAF-M2 共定位点的 L-R 网络得分显著高于 CAF/M2 主导点(Fig. 5f)。相比之下,CAF-内皮细胞共定位点与CAF/内皮细胞主导点之间没有显著差异(Supplementary Fig. 15c, d)。这些结果支持 CAF-M2 相互作用的预测,但不支持 CAF-内皮细胞相互作用。同时,使用不同反卷积方法估算的细胞类型比例,CAF-M2 相互作用均表现出持续显著性(Supplementary Table 4)。
通过整合收集的 scRNA-seq 数据集(Supplementary Table 2),作者还识别出几个预测的 L-R 对,可能介导当前乳腺肿瘤中 CAF 与 M2 型巨噬细胞之间的交互作用(Fig. 5g)。整合单细胞和空间数据的优势在于,可以排除两类假阳性:(1)具有空间邻近性但并非来自不同细胞类型的 L-R 对,因此它们不代表不同细胞类型之间的相互作用;(2)从单细胞数据中识别出但缺乏空间邻近性的 L-R 对,因此它们不代表细胞之间的物理接触(Supplementary Fig. 16)。
7. 肿瘤-免疫界面的细胞-细胞相互作用
SpaCET 的结果使得在空间背景下系统分析细胞间相互作用的生物学功能成为可能。例如,在乳腺肿瘤示例中识别的 CAF-M2 相互作用点显著靠近肿瘤-免疫界面(tumor-immune interface)(Fig. 6a, b)。因此,作者根据恶性细胞与存在 CAF-M2 相互作用的 ST 点的距离,将其分类为"近端(close)"或"远端(distant)"(Fig. 6c)。作者生成了近端与远端点之间的差异基因表达谱,并随后进行了基因集富集分析(GSEA)(Supplementary Fig. 17a)。远端的恶性细胞表现出细胞周期通路的富集,这是快速增殖癌细胞的典型结果(Fig. 6d)。相比之下,在近端恶性细胞中上调的基因主要属于上皮-间充质转化(EMT)通路(Fig. 6d and Supplementary Fig. 17b)。
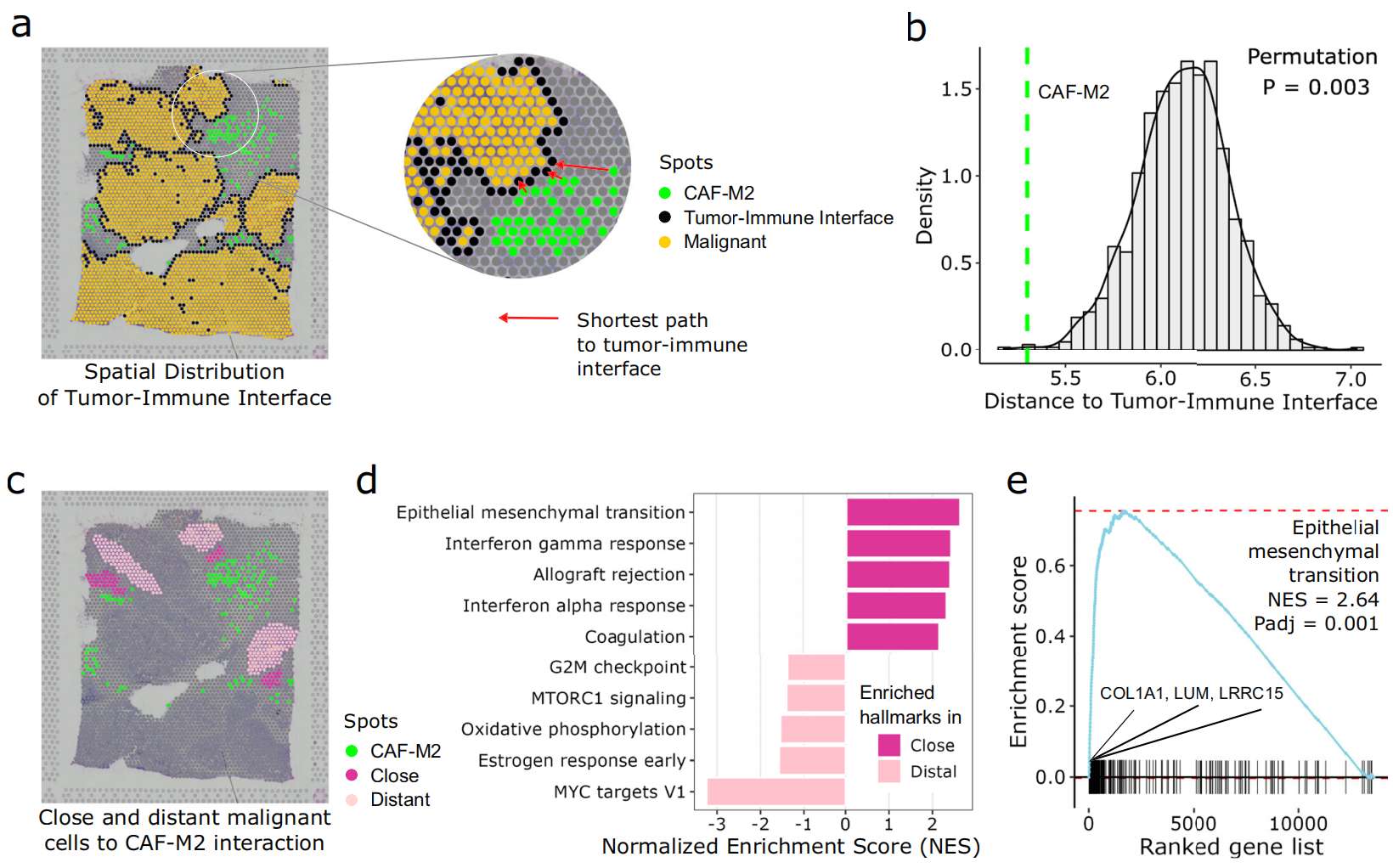 Figure 6. CAF-M2 相互作用与恶性细胞侵袭之间的关联
Figure 6. CAF-M2 相互作用与恶性细胞侵袭之间的关联
(A) 乳腺肿瘤示例中肿瘤区域与免疫区域交界处的 CAF-M2 点。CAF-M2 点到肿瘤-免疫界面的距离是该点到达界面的最短路径。
(B) CAF-M2 相互作用点与肿瘤-免疫边界的距离。绿线表示每个 CAF-M2 点到肿瘤-免疫界面的平均距离。距离的零分布通过 1000 次随机化计算得出。
(C) 相对于 CAF-M2 相互作用点的近端和远端恶性细胞点。
(D) 在控制恶性细胞比例的条件下,对近端与远端恶性细胞(相对于 CAF-M2 点)之间差异表达进行的基因集富集分析(GSEA)。
(E) 图 d 中上皮-间充质转化通路的 GSEA 富集图。X 轴表示根据图 d 中分析的差异表达排序的基因列表。沿 x 轴的黑色竖线代表该通路中的基因。如果某个通路的所有基因(竖线)倾向于富集在 x 轴的最左侧,则表明该通路在近端恶性细胞中活跃,反之亦然。青色线是该通路的富集曲线,红色虚线分别表示青色线的最大值和最小值。P 值通过双侧置换检验(n=1000 次随机化)计算,并采用 Benjamini-Hochberg 方法进行校正。
此外,GSEA 报告了 EMT 通路中的几个基因(例如 COL1A1、LRRC15 和 LUM),这些基因在靠近 CAF-M2 相互作用区域的恶性细胞中高表达(Fig. 6e)。已有研究表明,这些基因在乳腺癌及其他癌症类型中驱动癌症进展。例如,I 型胶原蛋白 α1(COL1A1)在乳腺癌细胞的细胞外基质中含量丰富,在癌细胞系中敲低 COL1A1 可抑制癌细胞迁移。Lumican(LUM)通过上调 MAPK 信号通路促进膀胱癌细胞的增殖和迁移。体外和体内研究表明,LRRC15 过表达可增强多种癌症类型(例如乳腺癌、骨肉瘤和软组织肉瘤)的转移能力。然而,仍需要额外的实验证据来验证这些基因在促进癌症侵袭性方面的生物学功能。
讨论
作者提出的 SpaCET 具有多种算法设计,以应对肿瘤 ST 数据中细胞比例反卷积的挑战。作者在具有广泛细胞分辨率的 ST 数据上,证明了其相对于现有反卷积方法的优越性能。SpaCET 基于一个涵盖多种肿瘤类型的拷贝数变异和表达变化的基因模式字典来估算癌细胞比例,该策略在预测准确性和运行效率方面均优于基于 inferCNV 的策略(Supplementary Fig. 13d)。这种基于字典的方法也应广泛适用于单细胞 RNA 测序数据分析中的恶性细胞识别,特别是对于传统 inferCNV 包无法处理的染色体稳定型肿瘤。
作者的分层分解策略限制了分解结果中同一亚谱系内密切相关细胞类型之间共线性的负面影响(Supplementary Fig. 2e)。先前的一个工具 MuSiC 也执行分层反卷积,但其需要用户根据 scRNA-Seq 数据的聚类结果手动定义细胞层次结构。作为优势,SpaCET 提供了一个汇总自多种肿瘤类型的全面分层细胞类型参考。此外,SpaCET 包含一个不可识别组分,以处理参考中缺失的细胞类型以及不同区域间的细胞密度变化(Supplementary Fig. 4d, e and Supplementary Fig. 13c)。
不同肿瘤区域的空间异质性是由肿瘤演化以及癌细胞与免疫/基质细胞之间的相互作用驱动的。尽管已有几种策略被开发用于在亚细胞分辨率的 ST 技术(例如 seqFISH+ 或 CODEX)中探索细胞间通讯,但由于基因覆盖度和分辨率的差异,它们不适用于多细胞(即点水平)ST 数据。SpaCET 能够结合共定位和配体-受体分析来研究多细胞 ST 数据中的细胞间相互作用。
SpaCET 存在若干局限性。第一,在估算恶性细胞比例时(Fig. 1b),ST 特异性的恶性表达谱是通过对肿瘤 ST 数据内所有已识别的恶性细胞点的表达谱进行平均生成的。因此,对于包含非常不同的癌细胞状态的肿瘤 ST 数据,恶性细胞比例的估算准确性可能会降低。第二,在反卷积过程中,作者假设参考细胞类型的标记基因在未知细胞类型中不表达。然而,作者的模拟分析表明,如果未知细胞类型中超过 30% 的现有参考标记基因出现高表达,会导致未知细胞比例被低估(Supplementary Fig. 18)。如果发生这种情况,有两种解决方法:(1)未知细胞类型可能来自与现有细胞类型相近的谱系,因此应归类为已知细胞类型而非未知类型;(2)用户应预先估算新组织中的现有细胞谱系。如果 SpaCET 的默认谱系未能全面覆盖输入样本中的细胞组成,用户可能需要添加额外的细胞特征,并使用定制的参考来运行 SpaCET。
SpaCET 是一个用于理解肿瘤中细胞空间组织以及空间组织如何影响癌症进展的框架。随着临床研究中空间转录组学数据的持续积累,我们预见 SpaCET 将为许多致癌过程提供机制性见解,并为当前抗肿瘤治疗的瓶颈提供解决方案。
--------------- 结束 ---------------
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。
