论文总结
1、有开源代码,本研究生成的数据和源代码存放在GitHub https://github.com/liuchuwei/PGLCN中,GitHub 使用Python和Pytorch实现。
2、对比方法仅和传统的机器学习方法进行对比
3、使用GNNExplainer进行生物学解释,整合TCGA中33种癌症类型的mRNA表达、CNV、DNA甲基化等多组学数据,结合Reactome通路层级关系构建患者特异性生物图。模型结构包含生物图构建模块、图学习与图卷积模块、以及基于GNNExplainer的图解释模块(生成特征掩码和边掩码)。
4、识别出关键通路子图,揭示Toll样受体(TLR)家族 和DNA修复通路与TMB状态密切相关,可作为联合生物标志物及潜在治疗靶点。结合胃癌免疫治疗队列(Kim cohort),进一步证实TLR信号通路和DNA修复通路与TMB及免疫治疗疗效的关联。
摘要
肿瘤突变负荷(TMB)已成为评估癌症免疫疗法疗效的重要生物标志物。然而,由于肿瘤本身的复杂性,TMB并不总是与免疫检查点抑制剂(ICIs)的反应性相关。因此,完善TMB的解释和情境化是提升临床结局的必要条件。本研究对TMB与33种人类癌症多组学数据之间的关系进行了全面调查。我们的分析显示,STAD、COAD和UCEC中TMB状态变化带来明显的生物学变化。虽然多组学数据为剖析肿瘤复杂性提供了机会,但从如此庞大的信息中提取有意义的生物学见解仍是艰巨的挑战。为此,我们开发并实现了基于通路相互作用信息的生物知情图神经网络PGLCN。该模型促进了患者按具有不同TMB状态的亚组分层,并通过增强可解释性,评估驱动性生物学过程。通过整合多组学数据进行TMB预测,我们的PGLCN模型优于以往传统机器学习方法,展现出更优的TMB状态预测准确率(STAD AUC:0.976 ± 0.007;COAD的抗击率:0.994±0.007;UCEC AUC:0.947 ± 0.023)以及提升可理解性(BA-House 评分:1.0;社区文学士:0.999;BA-网格:0.994;树周期:0.917;树格:0.867)。此外,PGLCN固有的生物学可解释性识别出Toll样受体家族和DNA修复途径,作为与胃癌TMB状态结合的潜在联合生物标志物。这一发现暗示了与胃癌免疫治疗潜在的协同靶向策略,从而推动精准肿瘤学领域的发展。
引言
免疫检查点疗法激活免疫系统,使免疫细胞能够识别并攻击肿瘤细胞。自美国食品药品监督管理局(FDA)批准了多种免疫检查点抑制剂(ICIs)以来,免疫疗法已经改变了多种癌症的治疗格局1,2。然而,只有部分患者能通过这些药物实现持久的完全缓解,凸显了预测性生物标志物和联合治疗策略的必要性。肿瘤突变负荷(TMB),用于衡量该数量癌症突变,最近逐渐受到关注。突变的积累可以产生新抗原,可能帮助免疫系统识别肿瘤3,4。然而,由于肿瘤复杂性,TMB并不总是与ICI的ICI响应性相关,因此需要对TMB进行解释和情境分析的细化5。多组学测序技术的最新进展加深了我们对肿瘤复杂性的理解6。从多组学信息中提取生物学洞见依然具有挑战性,但转化癌症基因组学中的预测模型解释对于为患者护理提供信息和阐明潜在的生物过程至关重要7。线性模型,如逻辑回归,往往提供更高的可解释性,但准确性较低。深度神经网络方法能够以强大的性能处理复杂结构和数据依赖,但其解释性较差。可解释性限制阻碍了深度神经网络在医学中的应用8。多种可解释性方法,如利用全局平均池处理图像问题的Grad-CAM9和解释黑箱分类器的Lime10,提升了我们对深度神经网络功能的理解。GNNExplainer 通过识别紧凑的子图结构和预测的关键节点特征,增强了图神经网络的可解释性11。本研究引入了一种新型生物学参考模型PGLCN,能够将癌症患者分层到具有不同TMB状态的亚组,并通过模型可解释性评估驱动性生物过程。我们利用集成的多组学数据(包括mRNA表达、拷贝数变异(CNV)和DNA甲基化12,在低维空间生成了多组学通路矩阵,并基于Reactome数据库建立了通路邻接关系。建模完成后,我们按照GNNExplainer实现的特征和边缘掩码进行了训练,以精准定位关键通路结构。大量分子剖析数据的可用性促进了对不同TMB状态的生物学机制的发现,这些机制与免疫治疗相关。通过分析33种癌症类型的多组学数据和TMB,我们观察到STAD、COAD和UCEC中TMB状态之间的明显生物学变化。最近的KEYNOTE-061临床试验发现TMB与接受pembrolizumab治疗的胃癌患者临床结局呈正相关13。我们进一步应用PGLCN方法预测胃癌患者的TMB状态。PGLCN模型的预测表现优于先前建立的模型。重要的是,通过可解释性,PGLCN揭示了Toll样受体家族和DNA修复途径可能作为与TMB的联合生物标志物,并可能作为胃癌免疫治疗的联合靶点策略14。
相关工作
近年来,深度学习在细致识别免疫疗法潜在受益者的有效性受到强调15, 16。两大策略被划定。第一个是通过多模态临床数据同化预测免疫疗法受益者,包括CT/MRI影像数据17、基因组序列18,19、免疫组化数据20。第二种策略在建模方法中融入了先验知识。目前,诸如TMB4、错配修复缺陷(MMR)21、肿瘤浸润淋巴细胞(TIL)22、MHCI\[23和PDL124等显著生物标志物在免疫治疗益处预后中发挥着关键作用。 研究人员正积极尝试将这些生物标志物与临床数据结合以增强建模16。例如,He 等人将TMB与CT影像结合,构建了针对高级非小细胞肺癌(NSCLC)的深度学习模型,最终形成了TMB放射学指标,展现出对ICI治疗响应性的深远预测能力25。然而,深度学习模型因其多元参数训练而固有的晦涩性,依然是一个挑战。**可解读的深度学习算法有望提升肿瘤免疫治疗研究的精度,并揭示新的生物学发现。深度学习可解释性技术可根据其基础原则在分类学上分为三种范式:基于模型的解释方法、基于影响的数学传播,以及利用先验知识的透明神经网络26。**前者通过神经元活动分析27或使用注意力机制28实现可解释性。基于影响的数学传播通过计算机突变内(ISM)前向传播机制29或基于梯度的反向传播12识别关键输入特征。近年来,鉴于深层神经元功能的复杂解读,出现了一个强调编码透明神经框架、并富含先前生物学知识的观点7,30。近年来,图网络在多个生物医学领域展现出无与伦比的性能优势31--37。例如,在基因组数据和组织病理数据并置时,丁等人证明了空间连通图模型能够实现准确的分子剖面预测32。在基因组学领域,Webber等人审视了图神经网络在分类多癌症类型中的能力37;在生物电信号处理领域,Duong等人将这些网络巧妙地分类心电图输出34。然而,对医学领域图网络可解释性的深入探索仍然大多未被充分利用。基于既有知识的图神经结构在发现关键生物调控网络方面具有潜力。
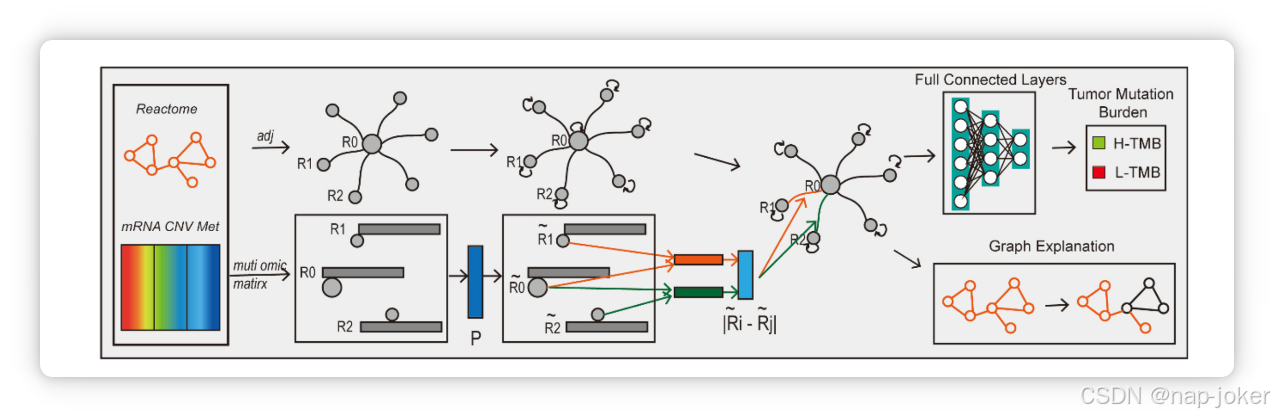
图1。可解释的生物知情深度半图神经网络。PGLCN由三个核心模块组成:生物图形成、神经网络和图解说。生物图形成模块基于生物先验和多组学数据构建患者特定的生物图。神经网络模块由图学习层、图卷积层和全连通层组成,能够学习新的通路相互作用模式并预测肿瘤突变负荷(TMB)状态。图解释模块生成特征和边缘掩码,以识别关键的生物子图结构,便于生成生物学相关假设。
方法
数据采集
癌症基因组图谱(TCGA)数据集来自UCSC数据库(http://xena.ucsc.edu/)\[38\]和cBioPortal数据库(http://cbioportal.org)\[39\]。获得了癌症患者的多组学数据(mRNA表达、CNV和DNA甲基化)及临床信息。简单核苷酸数据来自TCGA GDC(https://portal.gdc.cancer.gov/repository)。高肿瘤突变负定义为基因突变率\>百万碱基10个。采用了合成少数族裔过采样技术(SMOTE)来平衡数据。Reactome数据集来自Reactome通路数据集(https://reactome.org/)\[40\],包括通路、通路层级关系和通路基因集的完整信息。免疫治疗数据集来自潮汐数据库(http://tide.dfci.harvard.edu/download/)\[41--43\]。获得了金氏队列(胃癌免疫阻滞治疗队列)。合成数据集构建包括创建五个不同的节点分类数据集。BA-House 和 BA-Grid 的初始图基于基础的 Barab 'asi--Albert(BA)模式实现,包含 300 个节点。随后进行了80个网络图案的整合------具体来说,BA-House采用"house"五节点架构,以及附加在基础图中随机选取节点的3×3网格配置。BA-Community 数据集在表述中合并了两个 BA-House 图。在树循环和树格数据集的案例中,其基础结构是8层平衡的二叉树。随后,80个网络图案------包含树周期的6节点"环"结构和树格网格的3×3网格图案,融合到基本图架构中随机定位的节点11。
PGLCN结构概览
如图1所示,PGLCN包含三个主要模块:生物图形成、神经网络和图解释。对于每位患者,构建一个生物图,其中节点代表特定的通路,节点特征描绘每条通路的多组学特征,节点相互作用表示通路之间的关系。所提出的架构用于发现新的通路相互作用模式并提取特征。全连通层用于将分布式特征表示映射到标签空间,从而实现TMB状态预测。为了识别与TMB状态相关的关键子图,会训练一个独立模型用于模型解释,生成特征掩模和边缘掩模。
构造生物图
途径的层级关系来自反应组数据库,并构建了邻接矩阵A∈Rp×p,其中p代表反应组通路的数量。不同通路之间的直接相互作用标记为1,而没有直接相互作用标记为0。对角线元素设为1,以表示A中的自连关系。每条通路的多组学特征均基于mRNA表达、CNV和DNA甲基化数据构建,这些数据被转换为矩阵G、C、M∈Rn×r,其中n和r代表患者和基因的数量,且是循环的。对于每个反应组通路PI,从矩阵G、C和M提取相关基因,生成中间矩阵GI CI、MI ∈ Rn×ri,其中RI代表通路PI中参与的基因数量。主成分分析(PCA)被用于将GI CI和MI矩阵分解为无相关成分,得到Gpi、Cpi、Mpi∈Rn×q,其中q对应主成分(PC)的数量。该过程对所有反应组通路重复,生成合并矩阵 Gp、Cp、Mp ∈ Rn×pq,其中 p 代表反应组通路数量。重新排列和合并的矩阵三个矩阵中,Kp ∈ Rp×3q 被输入到 PGLCN 模型中,p 代表反应组通路的数量,列表示三种组学类型组合的 PC。例如,当q=2时,Kp由一个包含p×6个元素的矩阵表示,第一和第二列来自mRNA表达,第三和第四列来自CNV,第五和第六列来自DNA甲基化。
构建PGLCN的图学习模块
固定图可能不是图卷积神经网络的最优结构。此前已有研究提出GLCN等模型,该模型将图学习和图卷积整合成统一模型,以学习最优的图结构,以最佳服务于CNN44,45。Su 等人提出了一种图分类 CNN,用于识别远距离转移病例,嵌入图学习模块构建最优的蛋白质-蛋白质相互作用网络,从而最优地学习基因相互作用强度46。基于这些研究,我们设计了半监督图分类CNN,能够优化学习通路相互作用强度并预测患者的TMB状态。由于不同通路的复杂相互作用介导生物过程和细胞功能,Elmarakeby等人基于通路的层级关系构建了一个生物学知情的稀疏连通网络7。本研究基于通路关系和多组学数据,使用通路多组学矩阵 X = (x1, x2, ...xn) ∈ Rp×q 以及邻接矩阵 A = (x1, x2, ...xn)∈ Rp×p,其中p代表通路数量,n代表患者数,q代表主要治疗师数。我们构建了一个非负函数Sij = h(gi gj),以最优地建立通路的两对关系。路径矩阵的大维P降低了h(gi gj)在需要训练的长权重矢量时的效能。维数缩小通过以投影矩阵P ∈ Rnp×d, d < np为参数参数的低维嵌入网络实现。图学习是在单个层中构建的。
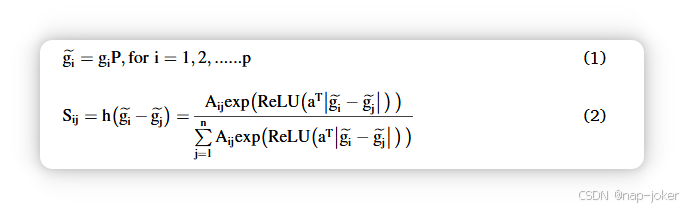
其中 ReLU(.) 是确保 Sij 非负性的激活函数,aT 是权重矢量。最终的软最大值操作确保所学图 S 能够满足以下性质:

也就是说,距离 ||GI GJ||2 鼓励较小的值 Sij。第二项是用来控制S稀疏度的正则化。
PGLCN的图卷积模块
如图1所示,PGLCN包含一个图学习层、多个卷积层和多个全连通层。由于图 S 满足以下性质:∑n j=1Sij = 1,Sij > 0,图卷积层被简化如下:

其中k = 0, 1, 2...k-1。X(k+1) 表示在 (k + 1)层激活的输出。σ,表示激活函数。W(k) 是一个可训练的权重矩阵。具体来说,W(0)∈ R1×g(0) 是一个输入到隐的可训练权重矩阵,W(k) ∈ Rg(k 1)×c 是一个隐藏到输出的可训练权重矩阵,其中 c 代表类号。交叉熵损失函数被最小化,以优化卷积层和全连通层中使用的权重矩阵。

其中Z ∈ Rn×2 代表预测标签,Y ∈ Rn×2 代表真实标签。整个架构的参数通过组合损耗函数进行了优化:

交叉验证
采用了5重交叉验证(CV)和5个重复验证来估算模型。采用分层K折叠法将样本分割。20%的样本被用作测试样品。其余数据进一步分为训练集(80%)和验证集(20%)。模型采用了准确性、回忆率、F1分数、AUC和精度来量化模型。该模型的性能与多个成熟的机器学习模型进行了比较,包括L2逻辑回归、(RBF)支持向量机、线性支持向量机、随机森林、自适应增强和决策树。
使用GNNExplainer进行生物学解释
GNNExplainer 是一种模型无关的方法,通过学习图掩码和特征掩码,选择重要子图,并掩蔽不重要的节点特征,来解释 GNN 在基于图的机器学习任务中的预测11。对于具有特征矩阵 X = (x1, x2, ...xn) ∈ Rn×q 和邻接矩阵 E = (x1, x2, ...xn) ∈ Rp×p,我们训练了一个图掩码M∈Rp×p,以及一个特征掩码F∈Rn×q,类似于GNNExplainer,用以计算通路的样本级重要性,其中p代表节点数,q代表基因组主特征的数量。我们的目标是识别一个子图Gs,特征为Xs = X ⊙ σ(F),相邻矩阵Es = E ⊙ σ(M),这对PGLCN的预测至关重要。我们利用互信息(MI)来选择最重要的结构和特征。
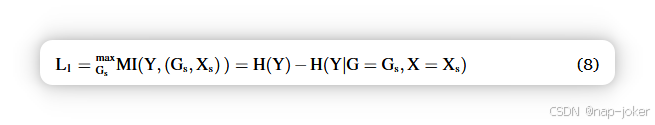
给定一个训练好的PGLCN模型Φ。MI量化了当特征矩阵限制为Xs、边矩阵限制为E时预测概率变化为"yy = Φ(G,X)。对于熵项 H(Y) 为常数,因为Φ在训练模型中是固定的,最大化互信息等价于最小化条件熵H(Y|G = Gs,X = Xs)。即:

结果
多组学生物特征的可视化
我们最初评估了33种人类癌症类型中不同TMB状态的生物学差异。每位患者的多组学生物学特征矩阵由 s 行(对应相同通路)和 3q 列(q 代表主成分数,即 PC)组成。在我们的模型中,q= 2,每种omic类型的前2个PC(按mRNA表达、CNV和DNA甲基化排序)构成了列。t-分布随机邻居嵌入(t-SNE)技术便于将高维数据映射到低维,同时保持局部特征。因此,我们利用t-SNE来可视化患者在不同癌症类型的多组学生物学特征(见图2)。值得注意的是,STAD、COAD和UCEC中不同TMB状态之间存在明显的生物学差异,使其区别于其他癌症类型。这一发现表明,高TMB水平的患者在这三种癌症类型中表现出保守的生物学改变。研究生物过程的具体变化可能提升TMB预测的准确性,并为免疫治疗策略提供新见解。
模型表现
随后,我们训练了PGLCN模型,用于对STAD、COAD和UCEC中高低TMB组进行分类。采用了5重验证(CV)方案,利用不同PC尺寸(1 5)(图3A)评估模型性能。拥有2台PC,PGLCN的性能达到饱和,STAD、COAD和UCEC的平均AUC分别为0.948±0.019、0.910±0.017,以及0.791±0.052。随后,我们试图确定模型中更具信息量的组学(见图3B-D)。首先,模型的性能是通过各个组学来评估的。在STAD中,PGLCN模型对每种组型的平均AUC分别为mRNA表达、CNV和DNA甲基化的0.976±0.007、0.900±0.013,以及0.976±0.004。此外,PGLCN模型结合两种组别型,mRNA表达与DNA甲基化组合的平均AUC为0.924±0.015、0.923、0.923±0.028、0.922±0.026,mRNA表达和CNV,以及CNV和DNA甲基化。COAD和UCEC也观察到类似结果。综合来看,这些发现表明mRNA表达对模型更具参考价值。
与传统机器学习模型的比较
我们采用t-SNE方法,研究从PGLCN最终全连通层提取的特征是否更具分离性(见图4)。经过图卷积运算后,这两个标签表现出高度可分离性。这一结果表明,PGLCN衍生的特征具有判别性,可能提升预测性能。此外,我们比较了PGLCN与六种不同机器学习方法的预测性能,包括L2逻辑回归、RBF支持向量机、线性支持向量机、随机森林、自适应增强和决策树。如表1--3所示,在STAD数据集中,PGLCN与线性支持向量机和随机森林模型相比其他方法展现出更优的预测能力。值得注意的是,PGLCN在回忆指标上达到了顶峰。在后续对COAD和UCEC数据集的评估中,PGLCn始终稳居首位。值得注意的是,PGLCN的标准差较小,显示出比其他方法更稳定的表现。
模型的可解释性
为检验PGLCN模型的可解释性,我们构建了五个不同的数据集,将GNNEXPLAINER作为参考基线。我们的发现强调了 PGLCN 在 BA-House、BA-Community、BA-Grid 和 Tree-Cycles 数据集中可解释性的提升。虽然 PGLCN 在树格网格数据集中相较于 GNNEXPLAINER 的准确性略有下降,但在识别关键子图配置方面表现出更明显的能力(见图 5A)。这些实证观察暗示PGLCN的半监督学习层增强了模型的可解释性。基于这一见解,我们利用PGLCN的可解释模块,挖掘出与胃癌TMB状态相关的显著生物网络结构。有趣的是,经过多次训练迭代,PGLCN能够熟练识别不同规模的生物子图。到了第200个训练阶段,PGLCN的学习轨迹接近饱和。分析第150纪元数据后,发现PGLCN倾向于以免疫为中心的疾病通路,重点关注托尔样受体(TLRs)信号相关通路(见图5B)。这些发现不仅暗示了TLR家族作为TMB组合免疫治疗预后指标的潜力,也使其成为针对不同TMB谱系的定制肿瘤-免疫干预的前瞻性治疗靶点。
TLR家族、胃癌肿瘤微环境与TMB之间的相互作用
TLRs是参与先天免疫调节的重要蛋白质分子类别。这些受体专门识别来自微生物的进化保守分子结构。在11个已识别的TLR家族成员中,有10个在人类免疫反应中发挥关键作用,部分成员位于细胞表面,另一些则局限于内体或溶酶体区室。最新研究将TLR与肿瘤发生的多个方面联系起来,包括免疫抑制、细胞凋亡和免疫系统激活47。我们的数据显示,大约19.22%的胃癌患者在TLR家族成员中存在突变。其中,TLR4表现出最高的突变患病率,占6%。相比之下,TLR2和TLR6的突变相对较少,仅占1%的比例(见图6A)。后续对不同TMB状态下的TLR表达模式分析显示,TMB升高患者中TLR3表达升高。相反,高TMB肿瘤中TLR4、TLR5、TLR7和TLR10表达减少(见图6B)。利用ssGSEA算法,我们辨别了肿瘤免疫细胞浸润的程度,并识别了肿瘤微环境特征。有趣的是,TLR4、TLR6、TLR8、TLR1、TLR2和TLR10与功能性免疫细胞浸润强度呈正相关。值得注意的是,这些TLR受体与肿瘤增殖动态呈反比关系(见图6C-D)。据报道,TLR作为连接先天免疫级联和适应性免疫级联的通道起到作用。通过促进细胞因子释放,它们可以增强细胞毒性T细胞,从而实现肿瘤湮灭和肿瘤负荷减轻。这些发现凸显了TLR作为联合肿瘤-免疫治疗的强大靶点的潜力,特别是针对多样化的TMB谱48,49。
与免疫治疗数据集的综合分析
为识别与TMB状态相关的生物学变化,这些变化对肿瘤免疫治疗具有潜在意义,我们采用了ssGSEA算法。这有助于在胃癌免疫治疗数据集中计算通路评分。利用Wilcoxon T检验,我们严格评估了与免疫治疗疗效相关通路。随后与 PGLCN 的图形解释集成,能够提取相关的子图结构。值得注意的是,PGLCN重点强调了核苷酸切除修复相关通路(图7A-B)。这些观察与既有的生物学知识高度契合。TMB升高与新抗原存在增加密切相关,促进淋巴细胞浸润增强。同时,表现出DNA修复缺陷的患者通常表现出较高比例的体细胞突变。这种关联关系密切相关高突变负荷与肿瘤修复相关通路的结合可能作为综合指标,预测肿瘤免疫治疗干预的疗效15,50--52。
讨论
免疫检查点阻断疗法可以激活患者的免疫系统,从而实现持久的反应。然而,这种疗法仅惠及一小部分患者,凸显了预测性生物标志物和联合治疗策略的必要性1,2。虽然肿瘤突变负荷(TMB)被认为是识别免疫检查点抑制剂(ICI)潜在反应者的宝贵生物标志物,但由于肿瘤复杂性,TMB并不总是与ICI反应性相关4。需要研究其潜在的生物机制并完善TMB的背景定位。多组学测序为肿瘤的基因组特性提供了见解,有助于解剖肿瘤复杂性,并在临床影响中发挥重要作用。基因组分析可能有助于完善TMB的情境化,并促进精准免疫治疗53,54。此外,综合基因组学优于单组学分析55,56。然而,从这些数据中提取具有生物学意义的洞察仍然充满挑战。本研究利用神经网络的高准确性和近期模型解释性进步,设计了基于生物信息的图神经网络------PGLCN------用于集成多组学数据分析。PGLCN提供了一个整合层级先验知识和多组学数据的框架,这些数据此前需要不同的统计方法。因此,PGLCN为每位患者构建独特的生物图,基于特定生物图预测TMB状态并识别关键子图结构。在33种人类癌症中,我们观察到STAD、COAD和UCEC中不同TMB状态间的基因组变化,表明存在共享的生物学变化。识别这些生物学变化有助于加深对TMB状态相关机制的理解。一项名为KEYNOTE-061的临床试验发现了TMB与接受pembrolizumab治疗的胃癌患者临床结局之间的正相关13。我们进一步应用PGLC预测STAD患者的TMB状态。PGLCN根据患者的生物图谱准确预测TMB状态,并识别出调控基因组不稳定性的关键生物结构。可视化生物图的重要子结构有助于发展关于癌症TMB状态所涉及的潜在生物过程的假说。此外,我们用胃癌免疫治疗队列评估了PGLCN43。值得注意的是,我们的分析明确指出TLRs相关信号图与胃癌患者TMB状态之间存在强烈关联。作为连接先天免疫与适应性免疫的关键通道,TLR已成为肿瘤学中,特别是肿瘤免疫治疗领域的有前景的治疗靶点47--49。这凸显了TLR作为肿瘤-免疫协同治疗中针对不同TMB状态的定制治疗靶点的潜力。在一项将PGLCN模型与免疫治疗数据集整合的平行研究中,PGLCN明显强调肿瘤修复相关通路。这种比对呼应了当时普遍的生物学理解,即DNA修复机制受损的患者往往表现出较高比例的体细胞突变。这些观察强化了高突变负荷与肿瘤修复中心通路之间的协同效应,可能作为预测肿瘤免疫疗法治疗成功的有力综合指标15, 50--52。尽管PGLCN提供了一个基于生物学基础的癌症基因组发现框架,但该模型在应用前需要培训。与所有深度学习模型一样,用于训练模型的超参数显著影响模型性能。此外,PGLCN基于生物先验构建生物图结构,使模型在不同任务中的应用依赖于这些先验。总之,PGLCN是一种生物学知情的图神经网络,能够准确分类具有不同TMB状态的胃癌患者。通过随机掩蔽识别重要的子图结构有助于生物学假设的生成。与免疫治疗队列的联合分析强调了NOTCH信号在肿瘤TMB状态适应和肿瘤免疫治疗中的关键作用。