前言
主要属于传统生物信息学的范畴,之前只懂原理,但是从来没有实操过
这次有机会做一下分析,记录一下完整过程
正文
主要分析流程:Salmon+tximport
Salmon安装
首先无root安装Salmon,先从github上下载:(太慢直接本地下载+scp)
bash
wget https://github.com/COMBINE-lab/salmon/releases/download/v1.11.4/salmon-linux-x86_64.tar.gz然后解压:
bash
tar -zxvf salmon-linux-x86_64.tar.gz把bin添加到.bashrc中的路径即可,在~/.bashrc中添加:
bash
export PATH=$PATH:/xxxxxxxxxxx/salmon-linux-x86_64/bin然后刷新一下bashrc
source ~/.bashrc
测试一下:
salmon -h,能看到指令提示说明安装成功
下载参考基因组数据
这个要看你分析什么东西了,如果分析蛋白质找编码区参考基因组,如果分析非编码RNA就找非编码的基因组。
进入NCBI的数据库
搜索你想下载的物种,例如Homo sapiens genome assembly GRCh38.p14 - NCBI - NLM
然后点download,弹出窗口,左边选择数据库,右边选择要下载的文件类型,例如我要下载编码区的文件,就勾选Genomic coding sequences
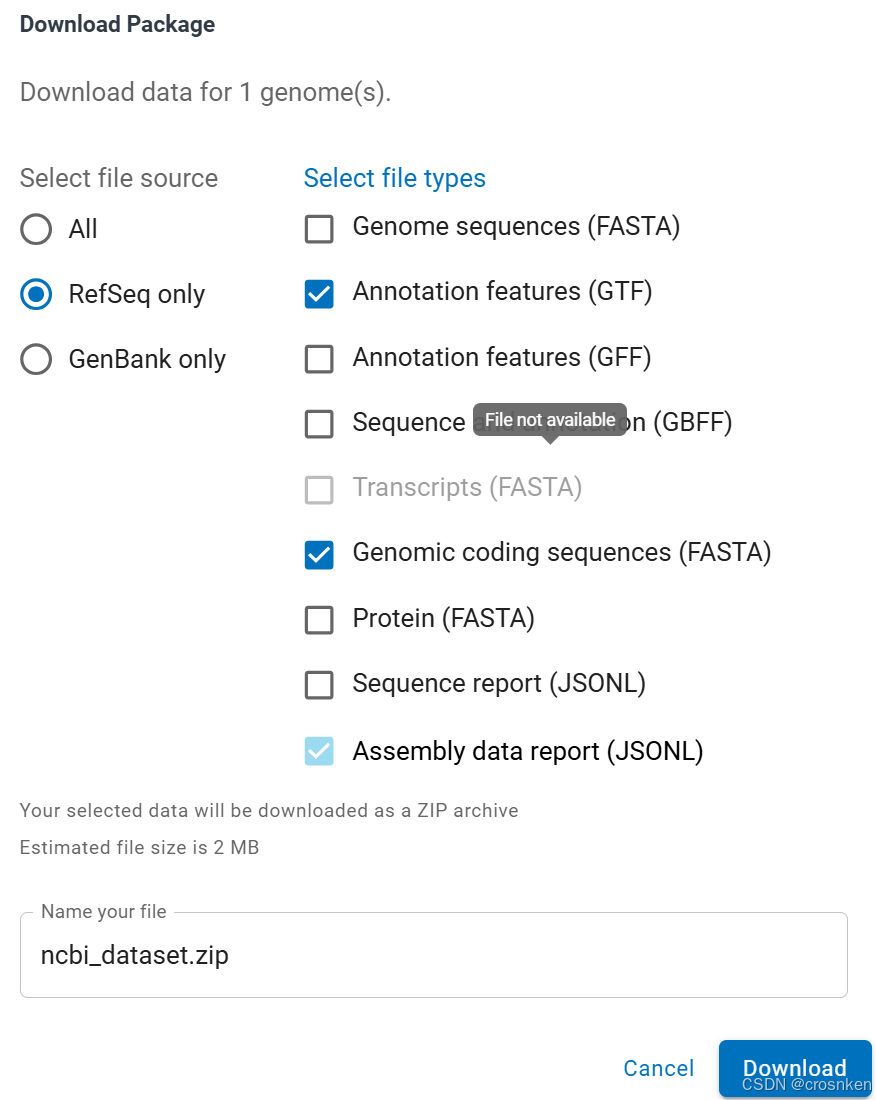
cds.fna的文件格式大概如下:
就是fasta格式,标注的基因编码区
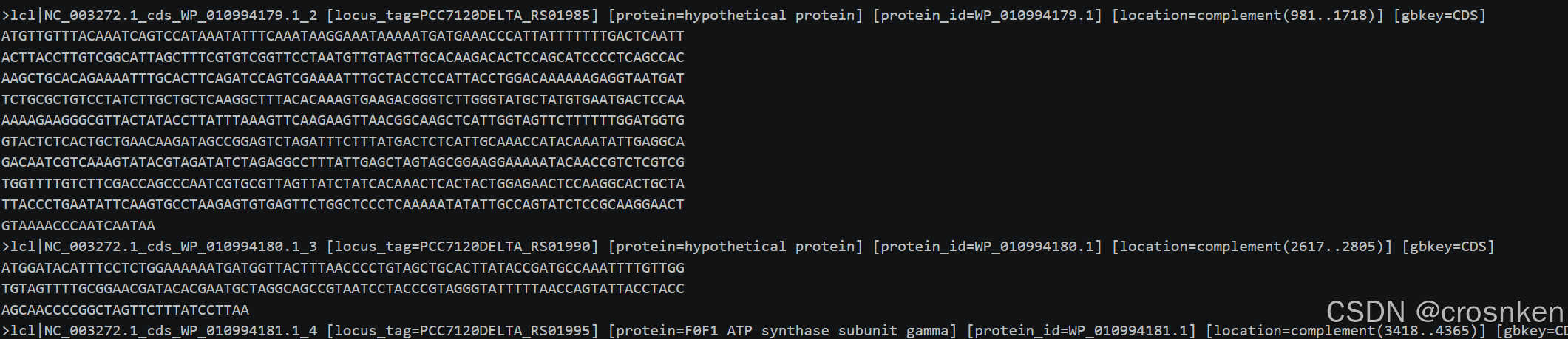
GTF的数据格式如下,它是对每个基因的标注,有启动子终止子以及编码区,可以看出来fna文件是标注的全基因(包含启动子和终止子)
Salmon分析
用salmon对下载下来的参考编码区建立索引
bash
salmon index -t cds_from_genomic.fna -i salmon_index -p 8然后跑脚本分析测得的采样数据,这个根据自己的路径和命名改一下就行。
bash
#!/bin/bash
INDEX="./salmon_index"
OUTPUT_DIR="quants"
mkdir -p ${OUTPUT_DIR}
for sample in "sample-1" "sample-2" "sample-3"; do
salmon quant -i ${INDEX} \
-l A \
-1 ./${sample}/${sample}.R1.fq.gz \
-2 ./${sample}/${sample}.R2.fq.gz \
--validateMappings \
-o ${OUTPUT_DIR}/${sample}_quant \
-p 8
done现代的分析很快了,大概不到三分钟就能分析十几个G的数据,而且我们只开了-p 8八线程分析。
出来的文件,一般只需要看quant.sf
.sf文件里面有五个指标
Name Length EffectiveLength TPM NumReads
前面两个分别是基因名称和长度,EffectiveLength是有效长度(能够有效进行序列比对的长度)
NumReads是Salmon估计的该基因包含的reads数量。
TPM = (NumReads / EffectiveLength) / Σ(NumReads_i / EffectiveLength_i) * 10^6
TPM用于衡量该段基因的相对丰度,因为长的基因占有reads的可能性更大(假设从均匀分布的角度来说),所以我们让基因的reads数除以该段基因的长度,得到该基因中平均每个位置占有的reads数。然后归一化,再乘以一百万,表示一百万个转录本中,该基因估计占用的转录本数量。
R语言环境构建
miniconda可以直接构建R语言环境
然后好像Mamba可以加速,效果确实不错,所以我们先给base环境装个mamba
bash
conda install -n base -c conda-forge mamba然后用mamba配R语言环境,装tximport和DESeq2
bash
mamba create -n renv -c conda-forge -c bioconda r-base=4.4 bioconductor-tximport r-readr bioconductor-deseq2 bioconductor-genomicfeatures然后激活环境,输入R,进入R语言终端,尝试引入tximport和DESeq2库
R
library(tximport)
library(readr)
library(DESeq2)
library(GenomicFeatures)没有报错就可以进入下一步
(其实我们也可以尝试给R语言的CRAN换源,但这个只在R语言命令行理使用install指令时需要用到,而这个会在第一次install时自动提示你选择镜像源,所以不用手动换)
在此之前,我们还需要安装一下txdbmaker,这个没法用mamba直接安装
于是我们使用R语言自带的包管理器:(在R终端执行)
install.packages("BiocManager")然后因为txdbmaker还依赖xml2,我们还需要安装r-xml2(在conda环境执行)
mamba install conda-forge::r-xml2
安装txdbmaker:(在R终端执行)
BiocManager::install("txdbmaker")(为了能够在vscode中使用R语言的相关插件,我们再安装一个langrageserver
mamba install conda-forge::r-fs
install.packages("languageserver"))
(也可以不装)
tximport构建基因表达矩阵
这里需要补一下R语言的txdb构建器,它读取GFF和GTF文件,构建一个小型的数据库,让用户通过select等函数来查询参考基因组数据。
数据库中主要包含以下几张表:
chrominfo、transcript、gene、cds、exon、splicing
分别表示染色体信息、转录本信息、基因、编码区、外显子、剪接位点
这里我们分析的是原核生物,暂时只看编码区的protein_id,以及对应的基因的old_locus_tag,然后转到.sf表中找到对应的表达量,就可以建立映射关系了。
找到cds和gene的关联可以通过,transcript_tx和cds_tx两个表来实现
上面的方法实在是太复杂了,干脆直接手动解析了。
首先处理以下之前的Salmon的输出,把第一列的protein_id提取出来,方便后续对齐:
R
sf_files <- list.files("quants", pattern = "\\.sf$", full.names = TRUE, recursive = TRUE)
for (sf_path in sf_files) {
sf_data <- read.table(sf_path, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
sf_data[, 1] <- sapply(strsplit(sf_data[, 1], "_"), function(x){paste0(x[4],"_",x[5])})
write.table(sf_data, paste0("salmon_out/",basename(dirname(sf_path)),".sf"), sep = "\t", row.names = FALSE, quote = FALSE)
}然后从GTF文件里面读取gene_id到protein_id的映射,与gene_id到old_locus_tag(KEGG id)的映射,这样我们才能再通过uniprot转到我们熟悉的uniprot ID上面。
R
library(tximport)
library(rtracklayer)
library(GenomicFeatures)
gtf_file <- "genomic.gtf"
gtf <- rtracklayer::import(gtf_file, format = "gtf")
gtf_df <- as.data.frame(gtf)
# 建立gene_id到old_locus_tag的映射
gene_rows <- gtf_df[gtf_df$type == "gene", ]
gene2olt <- unique(gene_rows[!is.na(gene_rows$old_locus_tag), c("gene_id", "old_locus_tag")])
head(gene2olt)
# 建立gene_id到protein_id的映射
cds_rows <- gtf_df[gtf_df$type == "CDS", ]
gene2prot <- unique(cds_rows[!is.na(cds_rows$protein_id), c("gene_id", "protein_id")])
head(gene2prot)
# 构建protein_id到old_locus_tag的映射
prot2olt <- merge(gene2prot, gene2olt, by = "gene_id", all.x = TRUE)[, c("protein_id", "old_locus_tag")]
prot2olt <- prot2olt[!is.na(prot2olt$old_locus_tag), ]
prot2olt <- prot2olt[!duplicated(prot2olt$protein_id), ]
head(prot2olt)接下来就可以用tximport构建基因表达矩阵了:
其实就是把几个样本的salmon结果的numreads拼起来
当然也可以更换countsFromAbundance的方法
R
# 使用tximport分析
sf_files <- list.files("salmon_out", pattern = "\\.sf$", full.names = TRUE)
names(sf_files) <- sapply(strsplit(basename(sf_files), "\\.sf"), function(x) x[1])
txi <- tximport(sf_files, type = "salmon", tx2gene=prot2olt)
# 输出结果
write.table(txi$counts, "gene_counts.txt", sep = "\t", row.names = TRUE, quote = FALSE)DESeq2差异表达分析
R
# 差异表达分析
library(DESeq2)
# 构建样本信息
sample_info <- data.frame(
sample = names(sf_files),
condition = c("A", "A", "A", "B", "B", "B", "C", "C", "C"),
stringsAsFactors = FALSE
)
rownames(sample_info) <- sample_info$sample
# 创建DESeqDataSet对象
dds <- DESeqDataSetFromMatrix(countData = round(txi$counts), colData = sample_info, design = ~ condition)
dds <- DESeq(dds)
res <- results(dds, contrast = c("condition", "A", "C"))
write.table(as.data.frame(res), "A2C_contrast.txt", sep = "\t", row.names = TRUE, quote = FALSE)这里contrast中,前面的A是实验组,后面的C是对照组
出来的表一共分为六列数据:baseMean log2FoldChange lfcSE stat pvalue padj
分别表示:样本表达量均值,log2FC=log2(A表达量/C表达量),log2FC的估计精度(标准误),
stat=log2FC/lfcSE(Wald检验统计量),pvalue = 2 × (1 - Φ(|stat|))(Φ是标准正态分布的累计函数,对正态分布不定积分),padj是对所有的基因的pvalue进行分布校正后的pvalue值(更准确)。
一般来说log2FC反映的实验组相比于对照组变化程度,padj<0.05就说明差异很显著。
WGCNA共表达分析
首先配一下环境依赖:
R
mamba install -c conda-forge zlib然后用R安装WGCNA:
R
BiocManager::install("WGCNA")在开始之前,需要补充一下基础知识:
皮尔逊相关系数:
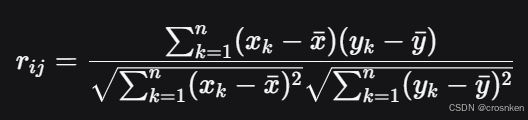
共表达网络分析算法流程
1、预处理
筛选出表达值较高的基因,然后取它们的对数
R
keep_genes <- rowSums(txi$counts) > 20000
datExpr <- txi$counts[keep_genes, ]
datExpr <- log2(datExpr + 1)2、利用皮尔逊相关系数构建邻接矩阵:
a_{ij}=|r_{ij}|^β,β是我们枚举的指数,r_ij是基因i和基因j在所有样本中的表达量的皮尔逊相关系数
由于共表达网络一般属于无标度网络,节点的度数服从幂律分布,所以我们可以通过枚举这个指数,计算边权(这里应该涉及了更深层的数学逻辑,为什么这样构造的网络能够符合节点度数服从β-幂律分布?)
然后计算TOM矩阵(拓扑重叠矩阵),k_i是节点i的度
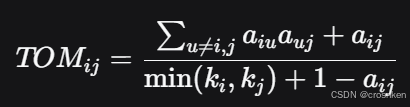
TOM_ij越大,表示这两个点越相邻,邻域重叠程度越大
R
# 转置表达矩阵,使基因为列,样本为行
datExpr <- t(datExpr)
# 构建共表达网络
powers <- c(1:10, seq(12, 20, by=2))
sft <- pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
softPower <- sft$powerEstimate
adjacency <- adjacency(datExpr, power = softPower)
TOM <- TOMsimilarity(adjacency)3、聚类UPGMA算法
我们使用1-TOM作为聚类使用的距离矩阵
熟悉最小生成树、Kruskal和Boruvka算法会觉得接下来的算法非常眼熟
UPGMA算法:
(1)初始每个点都是一个独立的集合
集合A,B之间的距离定义为:sum(dis(i,j) | 任意i∈A,j∈B) / (|A|*|B|),即A、B中所有点对距离的平均值
(2)选择距离最小的两个集合合并,更新合并后到其他集合的距离,这样的合并关系可以构成一棵Kruskal生成树
(3)直到只剩下一个集合,否则回到(2)
这样我们构成的一棵树,我们可以选择若干个不重叠的子树,只要能覆盖所有的叶子就可以作为一个聚类结果,接下来就是如何选取子树的问题。
cutreeDynamic:
(1)首先从叶子节点向上遍历,找到一个节点对应子树中的基因集合的内部距离显著低于到外部的距离,并且该子树包含的基因数不低于阈值,所处深度不高于阈值,则作为一个选定的子树。
(2)将剩下的没有被覆盖的节点以此计算与当前所有的选定子树的距离,如果到其中一个子树的距离差异足够显著,则将该节点划分到该子树。否则保留为灰色标记(grey)
(3)DeepSplit控制划分精度,取值为0~4,越大划分出的类越多。
虽然这部分内容细节很多,但是代码都是调库,反而显得很短:
还可以用plotDendroAndColors画图
R
dissTOM <- 1 - TOM
geneTree <- hclust(as.dist(dissTOM), method = "average")
dynamicMods <- cutreeDynamic(dendro = geneTree, distM = dissTOM, deepSplit = 2, pamRespectsDendro = FALSE)
dynamicColors <- labels2colors(dynamicMods)
table(dynamicColors)
png("gene_dendrogram_and_module_colors.png", width = 800, height = 600)
plotDendroAndColors(
geneTree, dynamicColors,
"Dynamic Tree Cut",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05,
main = "Gene dendrogram and module colors"
)
dev.off()4、共表达网络与表型的相关性分析
这个没什么好说的,就是单纯分析相关性,然后画一个热力图
R
# 共表达网络/基因与表型关联分析
MEList <- moduleEigengenes(datExpr, colors = dynamicColors)
MEs <- MEList$eigengenes
trait_dummy <- model.matrix(~ 0 + condition, data = sample_info)
colnames(trait_dummy) <- gsub("condition", "", colnames(trait_dummy)) # 清理列名
# moduleTraitCor <- cor(datExpr, trait_dummy, use = "p")
moduleTraitCor <- cor(MEs, trait_dummy, use = "p")
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nrow(datExpr))
write.table(moduleTraitCor, "module_trait_correlation.txt", sep = "\t", row.names = TRUE, quote = FALSE)
write.table(moduleTraitPvalue, "module_trait_pvalue.txt", sep = "\t", row.names = TRUE, quote = FALSE)
# 可视化关联热图
library(pheatmap)
png("module_trait_correlation_heatmap.png", width = 800, height = 600)
pheatmap(moduleTraitCor,
display_numbers = TRUE,
number_format = "%.2f",
main = "Module--trait relationships",
fontsize = 12)
dev.off()结语
大概弄了一天半,主要花的时间都在配环境上,熟悉之后就好了。
若有遗漏或者错误还请多多指正。