论文总结
-
研究任务
- 面向病理全切片图像(WSIs)+ 组学数据的多模态癌症生存分析,解决高维数据冗余、模态异质性、Transformer 效率低问题。
-
核心模型
- 提出ME-Mamba 多专家架构 ,由 3 个专家并行协作:
- 病理专家:处理病理图像,提取形态学关键特征。
- 组学专家:处理基因组数据,提取分子层面关键特征。
- 协同专家:负责跨模态特征融合。
- 提出ME-Mamba 多专家架构 ,由 3 个专家并行协作:
-
单模态专家创新
- 采用注意力引导 Mamba ,结合原始扫描 + 转置扫描 + 注意力重排扫描三分支并行。
- 显式聚焦高判别性实例,同时保留全局上下文,线性复杂度高效建模长序列。
-
跨模态融合创新
- 局部融合:用 ** 最优传输(OT)** 做 token 级细粒度对齐。
- 全局融合:用 ** 最大均值差异(MMD)** 做特征分布一致性约束。
- 融合后经 ** 双向 Mamba(BiMamba)** 进一步编码。
-
实验结果
- 在TCGA 5 个癌症数据集 上,C-index 平均超 SOTA 约8%。
- 相比 Transformer,显存占用降低 62.2%、计算量减少 65.2%,效率显著提升。
- Kaplan-Meier 与可视化验证模型可解释性强、风险分层精准。
-
核心贡献
- 首次将 Mamba 用于多模态生存分析,兼顾单模态关键信息与跨模态深度融合。
- 兼顾预测精度、计算效率、临床可解释性。
摘要
全幻灯片图像生存分析(WSIS)在癌症研究中至关重要。尽管取得了显著的成功,但病理图像通常只提供幻灯片级别的标签,这阻碍了从十亿像素的WSIS中学习区分表示。随着高通量测序技术的快速发展,结合病理图像和基因组数据的多模式生存分析已经成为一种很有前途的方法。然而,数据的高维性和模式之间的异构性给提取区分特征和有效融合模式带来了巨大的挑战。为了解决这些问题,我们提出了一个多专家Mamba(ME-Mamba)系统,该系统捕获有区别的病理和基因组特征,同时支持两种模式的有效集成。这种方法实现了互补信息融合,而不会丢失来自个体模式的关键信息,从而促进了准确的癌症生存分析 。具体地说,我们首先引入一名病理学专家和一名基因组学专家来分别处理单模态数据。这两位专家都是用Mamba架构设计的,该架构结合了传统扫描和基于注意力的扫描机制,允许他们从包含以下内容的长实例序列中提取可区分的特征大量冗余或不相关的信息。其次,**我们设计了一个负责通道融合的协同专家。它通过最优传输显式学习两个通道之间的令牌级局部对应,并通过基于最大平均差异的全局跨通道融合损失来隐式增强分布一致性。**然后,融合的特征表示被传递到曼巴主干以进行进一步的集成。通过病理学专家、基因组专家和协同学专家的协作,我们的方法以相对较低的计算复杂度实现了稳定和准确的生存分析。在癌症基因组图谱(TCGA)的五个数据集上的广泛实验结果证明了我们最先进的性能。我们将公开我们的代码。
引言
生存分析是临床预后研究和癌症预后评估的核心任务,旨在从特定的起点预测死亡或疾病复发等事件发生的时间,并准确评估患者的死亡风险。在临床决策过程中,它在加强诊断和告知治疗计划方面起着至关重要的作用1,2。对于癌症患者,病理图像和基因组数据等多模式数据提供了相互关联的关键信息,形成了患者分层和生存分析的基础3。然而,传统的生存分析方法往往依赖于短期临床指标和长期随访报告4、5、6、7,不仅耗时长,而且临床适用性有限。同时,癌症的复杂性要求对多样化和个性化的数据进行全面的评估,这给模型有效地捕获关键的区别性特征和整合数据异质性带来了巨大的挑战。因此,发展有效的特征提取和多模式融合方法成为构建稳健和自适应识别策划生存分析模型的关键和挑战。
近年来,随着深度学习技术的快速发展,医学图像分析取得了长足的进步。作为癌症诊断的金标准,病理图像越来越多地应用于生存分析8,9,10,11,12。病理图像直接提供了肿瘤细胞的微观形态特征和肿瘤微环境的信息。这种视觉特征与肿瘤进展、侵袭性和患者预后密切相关,为评估生存风险提供了形态学基础。然而,仅使用病理图像并不能捕捉到分子水平的生物信息。许多预后因素没有直接反映在形态特征上,仅通过图像分析很难揭示它们与生存结果的深层次相关性。这可能导致对肿瘤异质性的不完全解释。因此,整合病理图像和基因组数据的多模式生存分析方法13,14,15,16,17,18,19具有相当大的研究前景。为了准确地使用这两种模式进行生存分析,现有的大多数方法都使用基于Transformer的架构来实现跨模式交互并获得多模式表示。例如,一些方法使用基于Transformer的多实例学习来捕获全局信息14,或者将共同注意机制应用于通道融合19,从而从不同的角度获取互补信息。虽然这些方法在特征建模和跨模式交互方面表现出了很好的性能,但高维多模式特征很容易掩盖最初存在于单模式数据中的关键生存相关信息。这增加了过度适应与任务无关的功能的风险。此外,注意机制的二次计算复杂性导致在处理长序列或大规模多模数据时效率较低,经常导致忽略关键实例级特征。为了解决上述挑战,我们的目标是利用病理图像和基因组数据的单峰特征以及它们相互作用的多模式表示。这使得能够有效地捕获关键的每种模式固有的信息,同时通过集成的多模式功能促进全面的生存分析。此外,为了降低计算复杂度,我们探索了一种更高效的序列建模框架--Mamba,它以线性的计算复杂度保持了强大的建模能力。通过将选择机制和硬件并行算法集成到结构化状态空间模型(SSM)中,MAMBA在没有注意机制的计算负担的情况下有效地捕获了长范围依赖关系。MAMBA已被广泛用于信息社会世界首脑会议分类20、21、22和多模式融合23、24、25、26、27等任务。然而,现有的基于Mamba的多实例学习方法往往依赖于多个扫描方向来捕获实例之间的上下文关系,这可能不足以识别最具区分性的实例级别特征,如图1(A)所示。此外,目前基于Mamba的多通道融合方法往往只是简单地将不同通道的特征按顺序交织在一起,缺乏能够全面捕获跨通道关系的更深层次的交互机制。这一限制最终损害了所学习的跨模式表示的质量。在本文中,我们提出了一个多专家系统ME-MAMBA(多专家Mamba),它集成了病理图像和基因组数据用于生存分析。该系统并行处理包括病理学和基因组学在内的单峰数据以及它们的多峰融合。这种并行架构有助于深入理解影响单模式和多模式表示的生存结果的潜在因素。它在不丢失关键单峰信息的情况下实现了模式的互补积分,而基于MAMBA的结构显著提高了计算效率。更具体地说,该系统由两名单模式专家(一名病理学专家和一名基因组学专家)以及一名多模式融合的协同专家组成。单峰专家被设计为基于注意力的曼巴多实例学习模块。每位专家采用三种扫描策略。具体地说,注意力模型首先对序列中的每个实例进行评分,然后通过将注意力分数降至显式重新排序捕获有区别的关键实例。
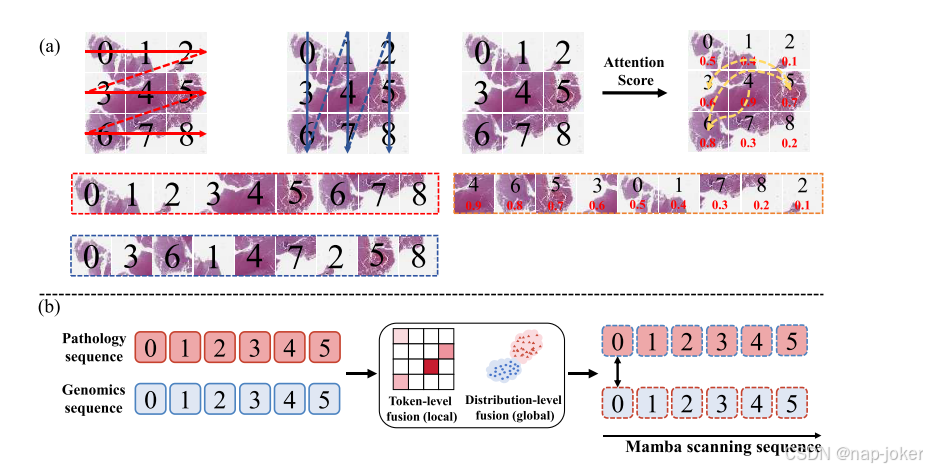
图1:每个专家模块的运行机制。(A)说明了单模态专家的不同扫描策略。传统的两种扫描策略直接从左到右和从上到下扫描实例序列,而基于注意力的扫描策略根据实例在序列中的关注度进行扫描。(B)多通道专家融合策略的说明。利用局部令牌级融合和全局分布级融合实现高效的多通道融合。
如图1(A)所示,这两个常规扫描方向用作捕获实例之间的全局上下文关系的补充。协同专家负责跨模式交互。我们首先使用最优传输来显式地学习两个通道之间的令牌级本地对应。应用基于最大均值差异的全局跨模式融合损失来隐含地增强分布一致性。然后,融合的表示被转发到曼巴主干以进行进一步的集成。这种多阶段融合策略确保了模型学习全面的多模式表示,如图1(B)所示。
通过病理学专家、基因组专家和协同学专家的合作,我们的方法有效地捕获了每种模式中的区别性信息,同时实现了彻底的多模式融合。本文的主要贡献可以概括如下:
·我们提出了一个多专家Mamba(ME-Mamba)系统,该系统首次实现了病理图像、基因组数据及其多模式融合的并行处理,有效地克服了基于Transformer的体系结构所固有的局限性。
·我们引入了一个注意力引导的Mamba架构(包括一名病理学专家和一名基因组专家),用于对病理和基因组模式进行建模。这种设计明确地捕获了每个单峰表示中的区别关键特征和全局上下文信息。
·我们开发了一个多通道曼巴架构(The Synergistic Expert),该架构融合了本地令牌级对齐和全球分布一致性,实现了全面的跨通道交互。
·我们评估了ME-Mamba在五个公共TCGA数据集上的性能,包括BLCA、BRCA、UCEC、GBMLGG、LUAD。在生存预测任务中,ME-Mamba取得了优异的性能,显著超过了所有比较方法(平均8%)。
相关工作
状态空间模型
状态空间模型(SSM)是一类序列建模框架,它通过潜在的状态转换来捕捉时间或序列依赖关系,使其特别适合于处理长序列。与Transformer中的自我注意机制不同,SSMS通过一系列潜在状态更新来对序列进行建模,实现了线性复杂性。S428通过稳定状态矩阵的对角化和通过柯西核简化计算,解决了早期SSM的高计算和存储代价。S529通过将多个独立的单输入单输出(SISO)SSM替换为多输入多输出(MIMO)SSM,消除了复杂的卷积核计算,进一步提高了效率。Mamba30(S6)通过结合依赖于输入的选择机制和硬件感知算法。与关注二次复杂性的Transformer相比,Mamba更擅长处理具有线性复杂性的长序列。利用SSMS的进步,人们对其应用于计算机视觉任务的研究兴趣激增。VIM31首先将Mamba引入计算机视觉任务,并提出了一种双向扫描策略来捕获上下文依赖关系。然而,由于扫描方向的限制,这种扫描策略无法捕捉到广泛的空间交互作用。后来,VMamba32设计了一个2D选择性扫描(SS2D)模块,它沿着四条扫描路径遍历图像块以扩大接收范围。EfficientVMamba33进一步引入了一种轻量级的交错扫描机制来逼近SS2D,从而降低了计算成本。在信息社会世界首脑会议的分析中,基于曼巴的方法也显示出显著的优势。MambaMIL20首先将Mamba框架与多实例学习相结合,用于WSI分析,除了原始扫描方向外,还通过从上到下重新排列序列来增强空间交互。为了进一步改善空间交互,MSMMIL22引入了网格扫描和层扫描策略,实现了在单个序列长度内的四向建模,并实现了高效的长序列建模。2DMamba21设计了一种新的2D扫描方法来直接处理2D特征地图,更好地保持了图像的空间连续性。然而,这些方法主要集中于获取全局序列信息和改善空间交互作用,而没有明确强调长序列中的区分性关键信息。相反,我们的方法引入了一种注意力引导的扫描机制,通过按照标记的注意力分数降序扫描标记来明确区分信息的优先级。结合传统的从左到右和从上到下的扫描方向,我们的方法高效地从长实例序列中捕捉到全局上下文关系和区分特征。
生存分析
**单模态。**生存分析是一种统计和建模方法,用于评估事件发生的时间和相关影响因素,可以为医生评估疾病进展的临床结果和治疗效果提供有价值的信息。传统的生存分析方法主要是单峰分析。在早期研究中,生存分析主要依靠COX比例风险模型34,利用短期临床指标和长期随访报告4,5,6,7,35。例如,Lai等人5将系统生物学和深度学习与15个生物标志物和临床数据相结合,以预测非小细胞肺癌(NSCLC)患者的生存结果。Yu等人35将基线预后参数与临床指标的动态趋势相结合,开发了急性/慢性肝功能衰竭(ACLF)患者的动态生存预测模型。病理图像直接显示肿瘤的形态特征,并提供有关肿瘤侵袭性、治疗反应和疾病复发可能性的有价值的信息。早期的研究侧重于病理图像分析,主要涉及分类36,37,38,39,40,41等任务,但也有一些工作也利用病理图像进行生存分析8,9,10,11,12,42。例如,WSISA8通过聚类和深度卷积网络使用病理图像实现了端到端的生存预测。DeepAttnMISL10使用了一种基于注意力的多实例学习(MIL)机制来聚合患者级别的表征,有效地对数据集进行生存分析。然而,单纯基于单峰病理图像的生存分析模型仍不能满足临床应用的需要。随着高通量测序技术的快速发展,基因组学数据已显示出作为疾病建模和预后指标的高度相关性43,44,45,46,从而为生存分析开辟了一条新的途径。
**多模态。**近年来,多通道数据融合在生存分析任务中得到了越来越多的关注。多模式数据在宏观、微观和分子层面提供对患者病情的多维洞察级别。通过整合来自不同模式的信息,可以实现对疾病的更全面的了解,从而导致更准确的诊断、优化的治疗策略和更可靠的预后预测。病理图像和基因组数据13,14,15,17,18,19,47,48,49的集成使得能够同时捕获肿瘤的形态特征和分子特征,从而增强了生存模型的预测能力。例如,MCAT19在WSIS和嵌入空间中形成的基因组特征之间提出了一种可解释的、密集的共同注意映射。CMTA14使用了两个并行的编解码器结构来分别处理病理和基因组数据,并使用跨通道注意模块作为桥梁来探索不同通道之间的关系并传递互补信息。CCL13设计了四个并行变压器编码器,将知识显式分解为四个组件,以实现更有效的多模式集成。现有的方法一般依靠交叉注意或自我注意机制来实现跨通道的交互和融合,旨在学习全面有效的多通道表征。然而,高维多峰特征往往掩盖了原始单峰数据中存在的与生存相关的关键信息。此外,Transformers的二次时间复杂性限制了其在处理大规模或长序列数据时的效率。这些限制突显了开发新的多模式融合方法以平衡有效性和效率的必要性。为了应对这些挑战,我们设计了一个基于Mamba的多专家系统,该系统并行处理单峰特征,并通过局部令牌级融合和全局分布比对获得多模表示,同时显著提高计算效率。
方法论
在这一部分中,我们首先描述多实例学习(MIL)和状态空间模型(SSM)的初步知识,然后概述所提出的多专家Mamba系统及其核心组件。
预定义
多实例学习
多实例学习(MIL)是一种弱监督学习方法,它将训练数据建模为包,其中每个包包含多个实例。如果包包含至少一个正实例,则将其标记为正。相反,一个袋子只有当它的所有实例都是负的时才是负的。例如,设xi={(xi,1,yi,1),(xi,2,yi,2),.。。,(xi,j,yi,j),.。。,(xi,n,yi,n)}表示第i个包,其中n为实例数。袋子的标签然后被定义为,

状态空间
受SSMS的启发,结构化状态空间序列(S4)模型已成为长序列建模的一种有效架构。作为线性时不变系统,S4模型由四个分量(∆,A,B,C)参数化,通过隐状态h(T)∈RN将一维输入序列x(T)∈RL映射到输出y(T)∈RL。这个过程可以用下面的连续系统来描述,


Mamba通过结合选择机制和硬件感知的并行算法进一步增强了S4模型。这使得该模型能够克服S4中静态参数的限制,通过依赖于输入的选择性传播或沿序列遗忘信息来实现有效的长序列建模。
概述和数据处理
框架概述
如图2(A)所示,所提出的多专家Mamba(ME-Mamba)系统由三个主要步骤组成:实例级特征提取、基于专家的特征处理和结果预测。在第一步中,每个病理全片图像(WSI)被划分为数千个互不重叠的斑块,而基因组数据则按功能类别进行分组。然后将特定的特征提取方法应用于每个通道,如第3.2.2节中所述。在第二步中,利用病理学专家和基因组专家分别从高度冗余的病理图像和基因组数据中提取可区分的特征。进一步使用协同专家来有效地整合病理和基因组特征。这些单元在第3.3和3.4节中有详细说明。最后,将提取的单峰特征和融合的多模态特征组合起来用于最终的生存预测,如第3.5节所述。
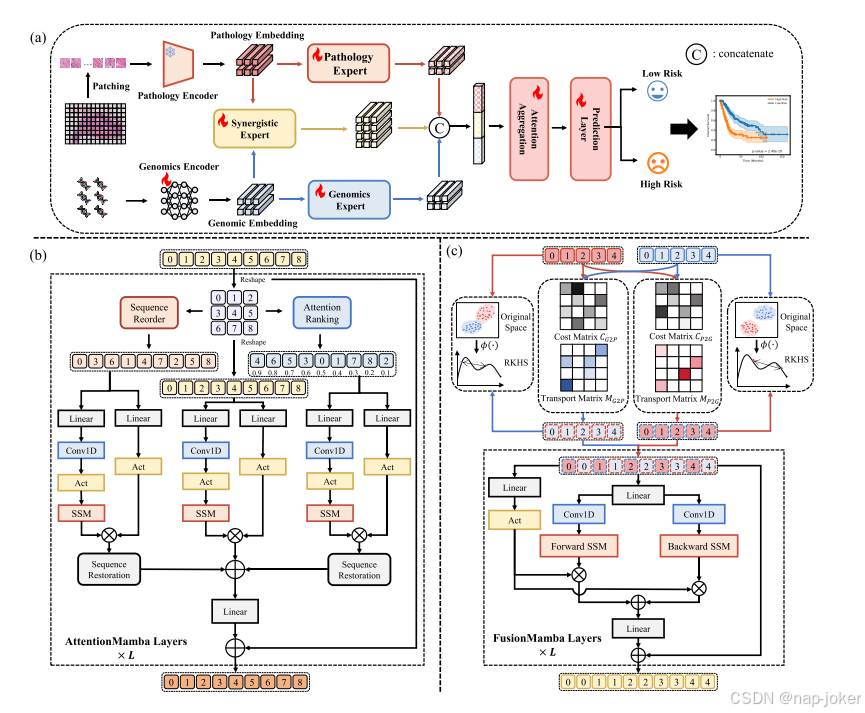
图2:(A)拟议的多专家Mamba(ME-Mamba)系统概述,该系统由病理学专家、基因组专家和协同专家组成。(B)病理学和基因组学专家架构示意图,该架构采用三种并行扫描方法,从长序列中捕捉全局背景和区别性特征。(C)协同专家的体系结构,包括特征融合、特征扫描和特征编码阶段。
特征提取
病理学。病理图像,即全切片图像(WSIS),提供了有关肿瘤免疫微环境的形态信息。然而,由于其极高的分辨率,WSIS不能直接被卷积神经网络处理,必须首先进行划分。我们首先分割组织区域,然后在20倍的放大倍数下提取大小为256×256的不重叠的斑块。在前人工作的基础上,我们采用了一个预先训练的ResNet5050模型,该模型最初是在ImageNet上训练的每个补丁的1024维嵌入。将同一WSI的所有补丁嵌入集合作为一个嵌入集。为了减少特征冗余和计算开销,我们使用多层感知器(MLP)将特征维度从1024降到256。然后,将得到的特征向量传递给病理专家Mamba模块以进行特征聚合。
基因组学。基因组学图谱可以识别与癌症预后相关的特定基因改变或生物标记物。某些基因突变、基因表达模式和DNA拷贝数变异可作为预后指标,有助于预测患者的生存结果。与以前的方法一样,我们将RNA序列(RNA-seq)、拷贝数变异(CNV)和简单核苷酸变异(SNV)序列划分为六个子序列13,14,18。利用两层自归一化神经网络(SNN)51将每个子序列变换为特征向量,然后应用多层感知器(MLP)获得256维表示。这些特征向量随后被传递到基因组学专家Mamba模块用于特征聚集。
病理学专家和基因组学专家
病理专家被设计为从病理图像中聚合实例级特征。它包括多个堆叠的基于注意力的Mamba层,这些层从包含大量冗余或无关数据的长实例序列中提取区别性特征和捕获全局信息,如图2(B)所示。通过将Mamba结构与多实例学习相集成,每个实例可以通过压缩的隐藏状态与先前扫描的实例进行交互,从而在降低计算复杂性的同时实现对长序列的有效建模。我们将传统的扫描策略与注意力引导的扫描机制相结合,形成了三分支并行的Mamba架构。传统的扫描包括两种策略:原始扫描和转置扫描。基于注意力的扫描机制根据实例的注意力分数对实例进行排序,使模型能够明确地专注于最具区别性的特征。
具体地,给定实例特征X∈Rn×d,其中n是序列长度,d是特征维度,我们首先将序列馈入三个并行分支。在第一分支中,保留原始序列顺序,并将其传递到后续的随机卷积和状态空间模型(SSM)层以进行序列建模。在第二个分支中,在MambaMIL20之后,实例序列被转置并顺序扫描,然后被相同的网络层处理。在第三个分支中,我们使用ABMIL52中的注意力机制为每个实例分配注意力分数。然后,实例按其注意力分数的降序进行重新排序,从而允许模型在后续处理中对高关注度的特征进行优先排序。总体程序制定如下:

基因组学专家遵循与病理学专家相同的计算过程,但使用不同的参数。通过这种方法,模型保留了原始序列的顺序和分布,并从全局视角,并强调通过基于注意力的重新排序来区分实例。这使得能够同时捕获全局上下文信息和关键实例级功能。
协同专家
Synergistic Expert旨在有效地集成来自病理图像和基因组数据的特征。它结合了两种互补的融合机制:基于最优传输(OT)的局部令牌级比对53和基于最大平均差异(MMD)的全局分布匹配54。这些都是双向应用的,通过使用病理学和基因组学特征作为锚。如图2(C)所示,产生的跨通道感知特征然后通过多个堆叠的多通道Mamba层来进一步增强融合。
局部跨模态融合
我们使用最优传输来实现两个通道之间的细粒度、令牌级对齐,并依次使用每个通道作为锚。该方法将特征序列视为离散分布,并学习将一个分布映射到另一个分布的成本最小化的传输计划,从而建立象征性对应。详细地,给定病理实例特征xp∈Rn×d和基因组实例特征xG∈Rm×d,其中n和m是序列长度,d是特征维度,我们的目标是学习一个捕获细粒度多模式关系的传输矩阵M。以病理学到基因组学(以基因组学为锚)的映射为例,目标制定为,
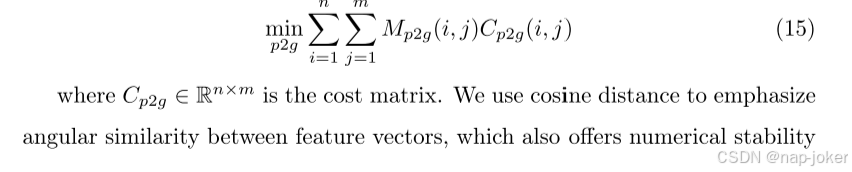


全局跨模态融合
为了隐含地对齐局部融合特征和锚定特征之间的全局分布,我们使用了最大平均偏差(MMD)。MMD通过比较不同模式在高维再生核-希尔伯特空间(RKHS)中的统计数据来衡量不同模式之间的统计差异。对于两个特征X和Y,MMD距离的平方定义为,
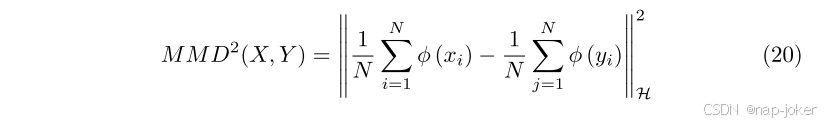
其中ϕ(·)表示从原始特征空间到RKHS H的映射。在实践中,我们使用核函数来计算该映射,

在训练过程中将这种损失降至最低,可确保融合后各模式之间的全局分布一致性。我们的策略结合了显式局部令牌级比对和隐式全局分布匹配,实现了多粒度比对,并在后续的Mamba骨干网中促进了更有效的特征编码。
多模态Mamba融合
在病理和基因组实例特征之间执行令牌级显式局部融合和分布级隐式全局比对之后,我们采用双向Mamba(BiMamba)31主干来进一步整合这两种模式。与传统的基于Transformer的方法通过自我关注同时处理所有令牌不同,我们的方法采用了有序扫描策略,在支持有效的跨模式交互的同时保留了Mamba的顺序性质。在给定局部和全局融合的特征序列的情况下,我们通过将两个模态的特征按顺序交织来构造统一的多模态特征序列,
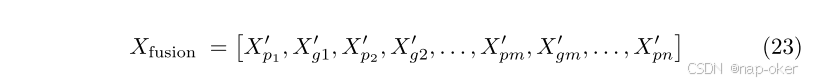
这种交错组织确保顺序处理来自不同医疗设备的要素,从而允许Mamba的选择性扫描机制有效地捕获模式内和模式间的依赖关系。多通道表示最终通过多个堆叠的BiMamba层获得。
特征聚合与预测
经过各自的专家模块处理后,得到病理实例特征序列、基因组特征序列和多峰特征序列,每个序列都包含有区分性信息。我们将这三个特征序列连接在一起,并将实例级特征聚合为BAG级表示。聚集方法遵循ABMIL52。最后,使用多层感知器(MLP)来预测危险函数H,

对于生存预测,我们在前人13,14,18,19,47,48的基础上,将原事件时间回归问题简化为一个分类问题。将患者的基本生存时间离散为n个相等的区间。事件发生的间隔tk被用作患者的分类标签k。该模型预测事件在每个时间间隔内发生的概率,形成危险向量危险向量H={h1,c,k}。每个患者样本被表示为三元组{H,c,k},其中c∈{0,1}表示正确的未审查状态。离散生存函数定义为fsurv(H,k)=qki=1(1−hi)。生存预测损失被表示为,

实验和结果
在这一部分中,我们在五个公共数据集上进行了广泛的实验,以评估我们所提出的模型的有效性。我们首先介绍了研究中使用的数据集和评估指标。然后,我们将实验结果与几种最先进的方法进行了比较,以证明我们的模型的优越性,并进行了可解释性分析。最后,我们进行了烧蚀实验,以考察关键部件的影响。
实验配置
**数据集。**为了验证该方法的性能,我们使用五个癌症数据集进行了一系列实验。这些数据集来自癌症基因组图谱(TCGA)1,其中包含成对的诊断完整幻灯片图像(WSIS)和基因组数据,以及来自数千名癌症患者的临床信息。其中膀胱尿路上皮癌(BLCA,n=372),乳腺浸润癌(BRCA,n=956),子宫内膜癌(UCEC,n=480),胶质母细胞瘤和低级别胶质瘤(GBMLGG,n=569),肺腺癌(LUAD,n=453)。对于WSIS,我们首先分割每张幻灯片的组织区域,然后以20倍的放大倍数将它们切割成256×256块。对于基因组数据,在前人工作13,14,19的基础上,我们使用了RNA-seq,CNV和SNV序列,并进一步将它们分成六个子序列:1)肿瘤抑制,2)肿瘤发生,3)蛋白激酶,4)细胞分化,5)转录,6)细胞因子和生长。
**评估指标。**采用一致性指数(C-index)57来衡量生存预测绩效。C指数衡量的是模型在生存分析中对个体生存时间进行准确排名的能力,评估预测的风险分数与实际生存结果之间的一致性。C指数可按如下方式表示,

**实施细节。**我们在五项癌症生存预测任务中使用了5次交叉验证来评估我们的模型和其他比较方法。具体地说,我们首先随机洗牌数据集,并将其分为五组,其中四组用作训练集,一组用作测试集。我们在训练集上训练模型,并在测试集上评估它们的性能,以报告相应的C指数得分Mean±STD(标准差)。我们采用了学习速率为1e-3的SGD优化器,并对该框架进行了30个周期的训练。所有实验都是在配备了NVIDIA GeForce RTX 4090图形处理器的平台上使用Python3.10和Pytorch工具包2.0版进行的。
与最先进的方法进行比较
为了验证ME-MAMBA方法的有效性,我们将其与单模态基线和多模态SOTA方法进行了比较。对于基因组数据,我们实现了SNN51和SNNTrans。对于病理数据,我们实现了ABMIL52、CLAM58、TransMIL59和DTFD60。对于多模式模型,我们选择了MCAT19、M3IF61、GPDBN15、Popoise62、HFBSurv63、SurvPath48、MOTCat18、CMTA14和CCL13。这些模型分为单模组和多模组,下面简要介绍几个具有代表性的模型。单模模型。对于基因组数据,SNNTrans是SNN51的变体,它使用自归一化神经网络(SNN)来提取实例级基因组特征,然后使用TransMIL将它们聚合到BAG级表示中。对于病理数据,ABMIL假设图像实例是独立的同分布(I.I.D.)并使用注意机制来聚集实例特征。TransMIL破解了身份识别。通过融合相关性建模和空间编码的假设,使用自我关注来聚合实例特征。多通道模型。MCAT使用共同注意机制在嵌入空间中动态对齐病理图像特征和基因组特征,然后使用Transformer进行多模式融合。然后,将聚集的特征串联以用于生存预测。SurvPath将转录数据分解成不同的生物路径签名,并使用稀疏注意转换器来模拟路径、病理图像和跨路径之间的交互。MOTCat使用最优传输来计算病理图像特征和基因组特征之间的全局匹配流,比传统的共同关注更有效地捕捉肿瘤微环境中的空间交互作用和基因共表达的结构一致性。CCL明确分解来自病理和基因组数据的交互知识,并在知识水平和患者水平上采用队列指导的监督。与单模模型进行了比较。如表1所示,所提出的方法在所有五个数据集上都取得了优异的性能。具体而言,它在BLCA上的C指数达到0.6993,比最好的单峰模型高8.3%;在BRCA上,C指数达到0.6910,表现出6.7%的改善;在UCEC上,C指数达到0.7063,提高2.4%;在GBMLGG上,C指数达到0.8669,超过最佳单峰模型3.6%;以及在LUAD上,0.7014,表现出10.7%的改善。这些结果表明,我们的方法有效地集成了多模式数据,并强调了多模式学习在生存分析中的好处。此外,我们观察到,使用基因组数据的单峰方法通常优于使用病理图像的方法,这表明基因组特征可能与患者生存结果显示出更强的相关性。我们的方法成功地利用了两种模式的互补信息,进一步提高了生存预测的准确性。
**与多模态模型的比较。**如表1所示,与现有最好的多模式方法相比,我们的方法也获得了更好的性能。具体地说,在最强的多模式基线上,它将BLCA上的C指数提高了1.2%,BRCA上提高了1.1%,UCEC上提高了0.5%,GBMLGG上提高了0.6%,LUAD上提高了0.8%。这一改进可以归功于我们的多专家体系结构,它在保留全局背景和支持有效的通道融合的同时,显式地捕获区别性信息。此外,与现有的基于Transformer的多模式方法不同,我们的方法利用了Mamba架构,从而提高了计算效率并减少了内存消耗。如图3所示,在不同的实例数下,我们将ME-Mamba与两种性能最好的基于Transformer的方法CMTA和CCL进行了比较。我们手动构建了1024维的实例向量,计数分别为1000、10,000和20,000,以模拟从WSIS提取的不同数量的图像块,同时将基因组特征组的数量保持在6个不变。首先,我们比较了不同实例数量的GPU内存使用情况(图3(A))。在所有设置中,Me-Mamba始终消耗最少的内存。在1000个实例上,ME-Mamba比CCL减少了39.4%的GPU内存使用量;在20,000个实例上,减少了62.2%,显示了其在处理大型全幻灯片图像方面的卓越效率。其次,我们分析了每种方法量化计算效率所需的FLOP(图3(B))。Me-Mamba在所有三个实例计数中实现了最低的FLOPS,证实了其高效率。在10,000个实例中,ME-Mamba与CMTA相比减少了65.2%的计算运算量,突出了Mamba架构在多模式融合任务中的优势,这与其较低的内存占用一致。为了进一步证明我们方法的稳健性,我们还使用了两个权威的基础模型Uni\[\]和Conch65来提取病理图像特征,并将结果与TOP多模方法进行了比较。我们的方法仍然具有很强的竞争力,并实现了最佳的整体表现。
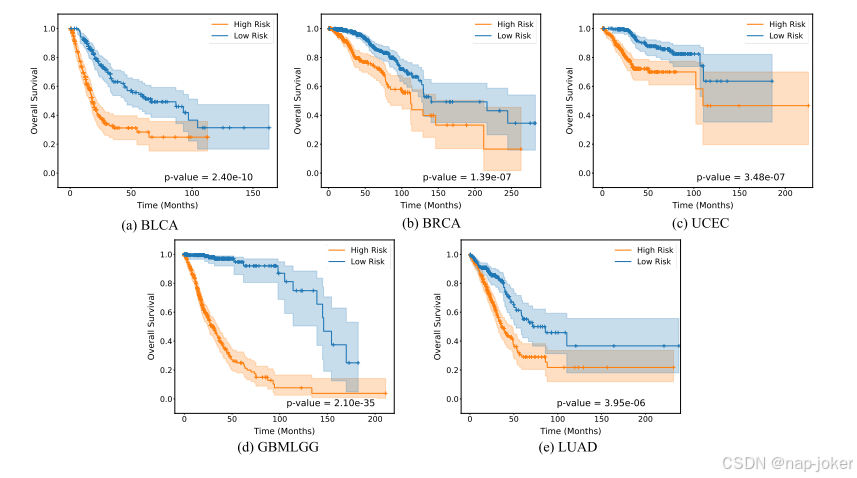
图4:Kaplan-Meier对五个TCGA数据集的分析,其中给出了低风险(蓝色)和高风险(红色)的患者分层。阴影区域指的是置信度区间。P值与0.05表示两组间差异有统计学意义,P值越低越好
卡普兰-迈耶分析
为了进一步验证ME-MAMBA用于生存分析的有效性,我们使用Kaplan-Meier分析来可视化所有患者的事件发生时间结果。Kaplan-Meier估计器是一种非参数统计方法,用于估计生存函数和分析事件间隔时间数据。具体来说,我们首先使用我们的模型预测每个患者的风险分数。然后根据中值风险得分将患者分为高风险组和低风险组。最后,我们使用Kaplan-Meier曲线来可视化生存事件,如图4所示。采用对数等级检验来评估高危(红色曲线)和低危(蓝色曲线)组之间差异的统计学意义。所有五个数据集的结果p值都显著低于0.05,表明我们的模型在生存分析中具有很强的区分力。我们还将我们的方法获得的p值与其他最先进的方法进行了比较,如表2所示。我们的方法在BRCA、UCEC和GBMLGG数据集上获得了最低的p值,同时在BLCA和LUAD上表现出了极具竞争力的性能。这些结果说明了我们的模型在多种癌症类型上的普适性和稳健性,表明它有可能通过可靠的生存预测来加强临床决策和癌症研究。
可解释性分析
WSI热图的可视化
我们在多专家系统中对病理专家进行了可解释性分析,以进一步展示其优越的性能,如图5所示。我们使用关注度权重来创建WISS上的热图,以突出具有代表性的病理斑块。具体地说,该模型根据每个图像块对最终预测的贡献来为每个图像块分配分数-分数越高表示越重要。我们将权重归一化在0到1之间(即,蓝色到红色),选择得分最高的6个块以获得详细的可视化,并分割细胞核。如图5所示,权重较高的斑块表现出相似的视觉特征,如核大小可变、核不典型、核浆比异常等,表明该模型可以自适应地聚焦于肿瘤区域,以帮助病理学家进行诊断。此外,来自不同风险水平患者的高质量斑块显示出不同的形态模式。例如,在低危病例"TCGA-3C-AALI"(存活时间:131个月)中,贴片呈现低度多形性。相比之下,高危病例TCGA-5T-A9QA(生存时间:9.95个月)显示出核严重拥挤的斑块。这些热图可视化表明,我们的模型可以有效地定位WSIS中的区分区域,这对于准确的生存预测至关重要。

要素的T-SNE可视化
我们使用T-SNE66来可视化病理专家、基因组专家和协同专家输出的特征分布。T-SNE最小化相似数据点之间的距离,最大化低维空间中不同数据点之间的距离。如图6所示,不同颜色的点代表不同专家的特征分布。这个特征集之间清晰的分离和最小的重叠强烈地表明每个专家模块在促进最终预测方面的互补作用。在每个专家内部,特征点被进一步聚为大约四个组,这与将地面事实生存时间划分为四个离散区间相一致。这种聚类行为证实了我们系统中的每个专家都有效地捕获了与生存预测相关的区别性模式。
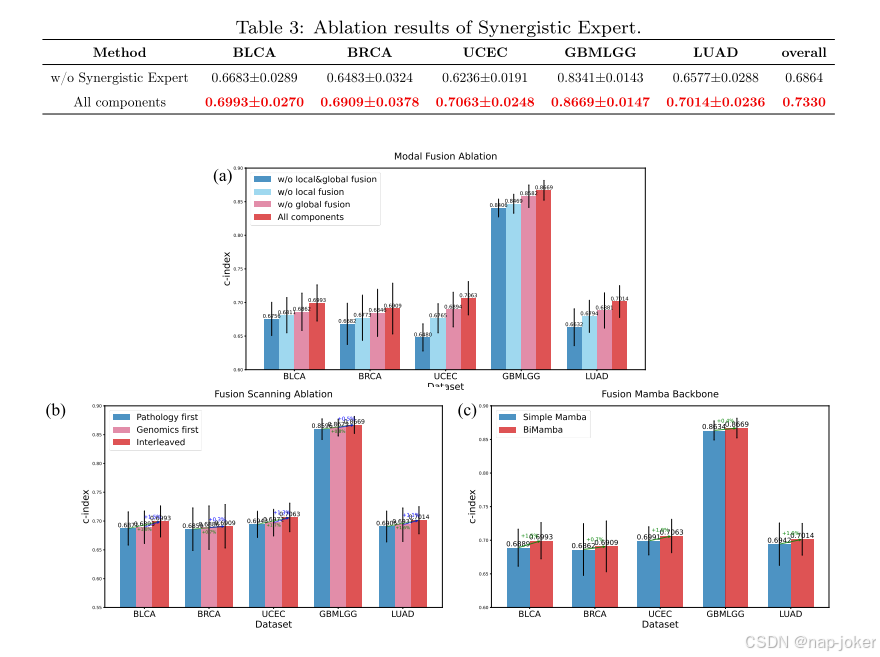
图7:协同专家三个阶段的消融结果。(A)特征融合阶段的消融。(B)特征扫描阶段的消融。(C)特征编码阶段的消融。
消融实验
协同专家的有效性研究
为了验证所提出的协同专家的有效性,我们进行了消融实验。首先,我们完全移除了协同学专家并对模型的性能进行了评估。如表3所示,所有五个数据集的性能都显著下降:C-index在BLCA上下降了4.4%,在BRCA上下降了6.2%,在UCEC上下降了11.7%,在GBMLGG上下降了3.8%,在LUAD上下降了6.2%。这些结果有力地证明了协同专家在跨模式融合中的重要性。接下来,我们保留了协同专家,并将其多模式特征处理分为三个阶段:特征融合、特征扫描和特征编码。在特征融合阶段,我们评估了局部跨模融合和全局跨模融合的贡献。结果如图7(A)所示。移除这两个组件中的任何一个都会导致性能下降。例如,在UCEC数据集上,去除局部融合会使C指数降低4.2%,而去除全局融合会导致2.4%的降低。值得注意的是,与相反的情况相比,当局部融合被保留而全局融合被移除时,模型表现得更好。这可以归因于这样一个事实,即局部融合通过最优传输显式地对齐各通道之间的令牌,而全局融合使用MMD隐式地匹配特征分布。在特征扫描阶段,我们比较了提出的两种模式的交错扫描和两种不同的扫描策略:(1)先扫描病理特征序列,然后扫描基因组序列。(2)先扫描基因组特征序列,然后扫描病理序列。如图7(B)所示,这两种替代策略在所有数据集上的表现都逊于交错方法。这是因为顺序扫描导致模型一次主要集中在一种模式上,限制了有效的融合。此外,先扫描基因组特征比先扫描病理特征导致的性能下降更小,这与单峰基因组方法通常优于单峰病理方法的观察结果一致,这可能是因为基因组数据与生存结果的更强相关性。在特征编码阶段,我们将提出的BiMamba骨干网与一个简单的MANBA模型进行了比较。图7(C)中的结果表明,BiMamba实现了更高的c指标值,这是由于其双向建模能力,它既处理前向序列上下文又处理后向序列上下文,并提高了密集预测任务的性能。
病理学专家和基因组专家的有效性
我们的病理专家和基因组专家Mamba模块包含三种扫描策略:原始扫描、转置扫描(共同构成传统扫描策略)和建议的注意力引导扫描。为了评估这些多重扫描机制的有效性,我们设计了三种实验设置进行比较:只使用原始扫描,同时使用原始扫描和转置扫描,以及同时使用原始扫描和注意力引导扫描。实验结果汇总在表4中。实验结果表明,与单独使用原始扫描相比,使用两个或更多扫描策略可以持续提高性能。这一增强归功于多个扫描策略能够更全面地捕获所有实例之间的关系。此外,我们观察到,将原始扫描与建议的注意力引导扫描相结合会产生比原始扫描和转置扫描相结合的更高的性能。这是因为根据注意力得分对实例进行排序,使模型能够更多地关注与生存结果高度相关的区别性实例。然而,仅使用两种扫描策略仍不能达到最佳性能。当这三种策略结合起来时,效果最好。这种集成的方法使模型能够同时捕获与生存和全局上下文关系相关的区别性实例级信息,而原始扫描补偿了可能被其他两种策略忽略的特征细节。
总结
在本文中,我们提出了多专家Mamba(ME-Mamba),这是一个多模式生存分析的开创性系统,通过三位专门的专家协同病理图像和基因组数据。这代表着在将Mamba架构应用于多模式生存分析任务方面取得了重大进展。在我们的系统中,病理专家和基因组专家使用一种注意力引导的扫描机制来从十亿像素的WSIS和高维基因组数据中提取区别性特征,显式地捕获关键实例级别的信息,同时保留全局上下文。协同专家将基于最优传输(OT)的局部令牌融合和基于最大平均偏差(MMD)的全局分布匹配相结合,以全面模拟跨通道交互。由此产生的表示通过BiMamba主干进一步精炼,以产生丰富的多模式特征。在五个TCGA数据集上的广泛实验验证了ME-MAMBA的最先进的性能、高的计算效率和强大的临床可解释性。鉴于其强大和可推广的性能,所提出的模型可以在未来扩展以集成更多的数据模式,为其适应涉及不同数据类型的更复杂的任务铺平道路。