文章目录
- [Notes 课件](#Notes 课件)
-
- 为什么我们需要统计力学视角?
- 先简单回顾一下我们的统计学知识(概率论与数理统计)
-
- 概率基础:采样与直方图
-
- [1. 随机过程与采样](#1. 随机过程与采样)
- [2. 绝对概率 vs 相对概率](#2. 绝对概率 vs 相对概率)
- 概率分布类型
-
- [1. 离散均匀分布(公平骰子)](#1. 离散均匀分布(公平骰子))
- [2. 离散有偏分布(作弊骰子)](#2. 离散有偏分布(作弊骰子))
- [3. 连续概率分布(高斯分布)](#3. 连续概率分布(高斯分布))
- 核心统计量
- 多维概率分布
-
- [1. 投影(边缘分布)](#1. 投影(边缘分布))
- [2. 相关性](#2. 相关性)
- 核心桥梁:玻尔兹曼分布
-
- 什么是玻尔兹曼分布?
- 玻尔兹曼分布与softmax的关联
- 统计与力学的连接:玻尔兹曼分布
-
-
-
- 1.核心公式来源:相空间概率拆分
- [2. 拆分后两个边缘分布](#2. 拆分后两个边缘分布)
-
- [1. 关键结论](#1. 关键结论)
- [2. 分子动力学意义](#2. 分子动力学意义)
-
- 各态历经性(Ergodicity)/遍历性
- 自由能:宏观态的有效能量
-
- [自由能 F i F_i Fi定义:从微观势能 U ( x ) → U(x)\to U(x)→宏观状态有效能(微观态积分粗粒化)](#自由能 F i F_i Fi定义:从微观势能 U ( x ) → U(x)\to U(x)→宏观状态有效能(微观态积分粗粒化))
-
- [1. 微观态 vs 宏观态(State)](#1. 微观态 vs 宏观态(State))
- [2. 宏观态占据概率(积分玻尔兹曼权重)](#2. 宏观态占据概率(积分玻尔兹曼权重))
- [3. 自由能定义式(关键公式)](#3. 自由能定义式(关键公式))
- [1. 状态(State)的定义](#1. 状态(State)的定义)
- [2. 自由能与概率的关系](#2. 自由能与概率的关系)
- [能量 - 概率 - 时间的统一关系](#能量 - 概率 - 时间的统一关系)
-
- [1. 微观与宏观的分野](#1. 微观与宏观的分野)
- [2. 时间尺度与自由能垒](#2. 时间尺度与自由能垒)
- [微观规律↔宏观规律总结(能量 U U U vs 自由能 F F F;概率指数依赖能量差)](#微观规律↔宏观规律总结(能量 U U U vs 自由能 F F F;概率指数依赖能量差))
- 溶液中分子的随机动力学图像
-
- [生物大分子随机模型(符号从 k B T → R T k_BT\to RT kBT→RT; F → G F\to G F→G吉布斯自由能,摩尔热力学)](#生物大分子随机模型(符号从 k B T → R T k_BT\to RT kBT→RT; F → G F\to G F→G吉布斯自由能,摩尔热力学))
- 3组核心公式
- 符号差异汇总
- 整体理论逻辑链
- 生物分子过程实例
- 结构生物信息学统一纲领(收尾逻辑)
-
- [两大永恒核心问题:采样(Sampling) + 能量(Energy/打分Score/损失Loss)](#两大永恒核心问题:采样(Sampling) + 能量(Energy/打分Score/损失Loss))
-
-
- 一、底层物理逻辑:玻尔兹曼分布是二者的数学桥梁
- [二、一一对应:物理能量 ↔ 打分Score / 损失Loss(结构生信&AI通用等价关系)](#二、一一对应:物理能量 ↔ 打分Score / 损失Loss(结构生信&AI通用等价关系))
-
- [1. 分子对接、传统结构预测:势能=Score打分函数](#1. 分子对接、传统结构预测:势能=Score打分函数)
- [2. 蛋白质AI结构预测:自由能=Loss损失函数](#2. 蛋白质AI结构预测:自由能=Loss损失函数)
-
- 如何从统计物理的角度来理解深度学习?
- [Notebooks 练习](#Notebooks 练习)
-
- 课堂练习
- Task
- Report
-
-
-
-
- [1. 把直方图 → 自由能图](#1. 把直方图 → 自由能图)
- [2. 定义状态](#2. 定义状态)
- [3. 计算核心物理量](#3. 计算核心物理量)
- 第1步:把「计数」变成「概率」
- [第2步:概率 → 自由能(核心公式)](#第2步:概率 → 自由能(核心公式))
- 第3步:自由能归一化
- [第4步:定义状态 → 算能量差/布居数](#第4步:定义状态 → 算能量差/布居数)
-
-
-
Notes 课件
从这一节课开始,我们迅速从实验理论部分转向生物分子动力学的统计力学视角,

纵观课件全文,我们只需要知道一点:
统计和力学之间是有联系的,关键在于玻尔兹曼分布,以及能量(energy)的概念,
另外核心(我个人总结)就是概率分布已经隐含了能量景观的信息。
为什么我们需要统计力学视角?
答案很简单:
- 统计:一大群分子构成的1个复杂体系
- 力学:分子之间相互作用与运动

生物分子系统具有三个本质特征:
- 可以改变自身形状(构象变化)
- 在三维空间中持续运动
- 分子间 / 分子内存在复杂相互作用
要描述这样的系统,我们需要:
- 力学:解释分子如何运动、如何相互作用
- 统计学:处理海量原子、海量构象的集合行为
生物分子解读:
我们永远无法追踪单个原子的精确运动轨迹,但可以通过统计方法描述构象的分布规律------ 这正是 X 射线晶体学给出平均结构、分子动力学给出构象集合的理论依据。
但是力学和统计学的观点并不是分立的,在我们的研究对象中,它们是可以交汇联系起来的,
而这个桥梁,就是能量(energy)的概念。
我们学习基础工科物理,无论是中学还是大学本科,基本上都是从运动学,深入到动力学,
先学简单的动力学,再深入刚体动力学,
而动力学的终点,基本上就是各种能量的表征和转化,
运运动学和动力学的学习终点,基本上就是用能量统一描述系统所有运动、相互作用;而统计物理,则在能量的基础上,从微观个体运动过渡到宏观集合行为,实现力学规律与统计概率的双向锚定。

所以,在研究分子体系中,关键在于,如何用力学里的「能量」概念,去解释统计学观测得到的概率规律 ?
先简单回顾一下我们的统计学知识(概率论与数理统计)
这一块基本上就是本科工科数学基础知识:概率论与数理统计,
我之前也总结过一些系统的课程大纲,可以参考:概率统计baseline
概率基础:采样与直方图

1. 随机过程与采样
- 随机(Stochastic) :事件由概率决定,通过大量观测可以得到不同结果的概率分布
- 采样(Sampling):通过多次观测统计结果,用直方图估计概率分布(分子动力学模拟的本质就是对构象空间的采样)
2. 绝对概率 vs 相对概率
| 类型 | 公式 | 含义 | 生物分子应用 |
|---|---|---|---|
| 绝对概率 | p ( i ) = count ( i ) ∑ k count ( k ) p(i) = \frac{\text{count}(i)}{\sum_k \text{count}(k)} p(i)=∑kcount(k)count(i) | 某一结果出现的真实概率,总和为1 | 某一构象在整个构象空间中出现的概率 |
| 相对概率 | p ( j ) p ( i ) = count ( j ) count ( i ) \frac{p(j)}{p(i)} = \frac{\text{count}(j)}{\text{count}(i)} p(i)p(j)=count(i)count(j) | 两个结果出现概率的比值 | 比较两个构象的稳定性(比值越大,构象j越稳定) |
关键结论:很多时候我们只需要相对概率就能比较构象稳定性,这大大降低了计算难度。
概率分布类型
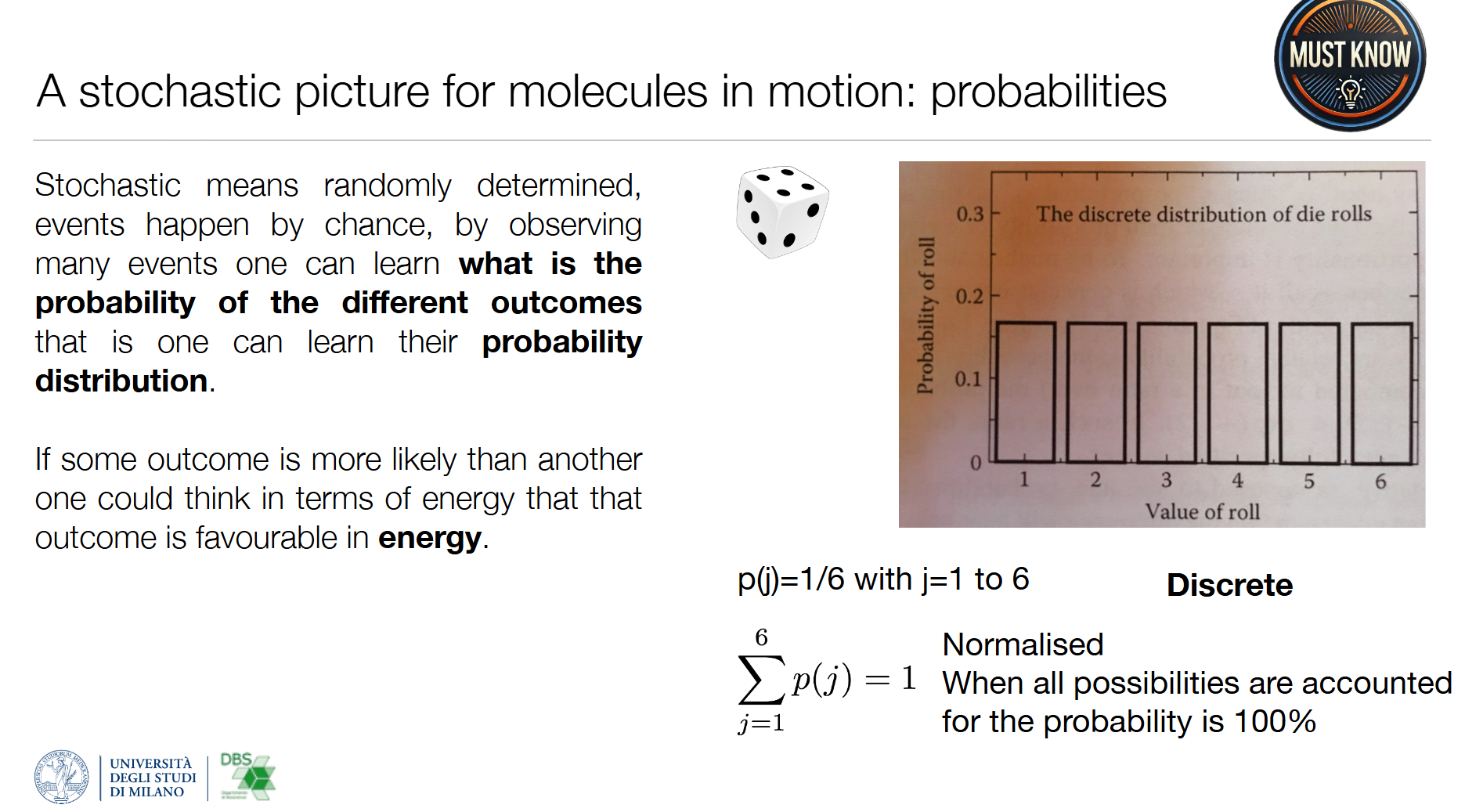
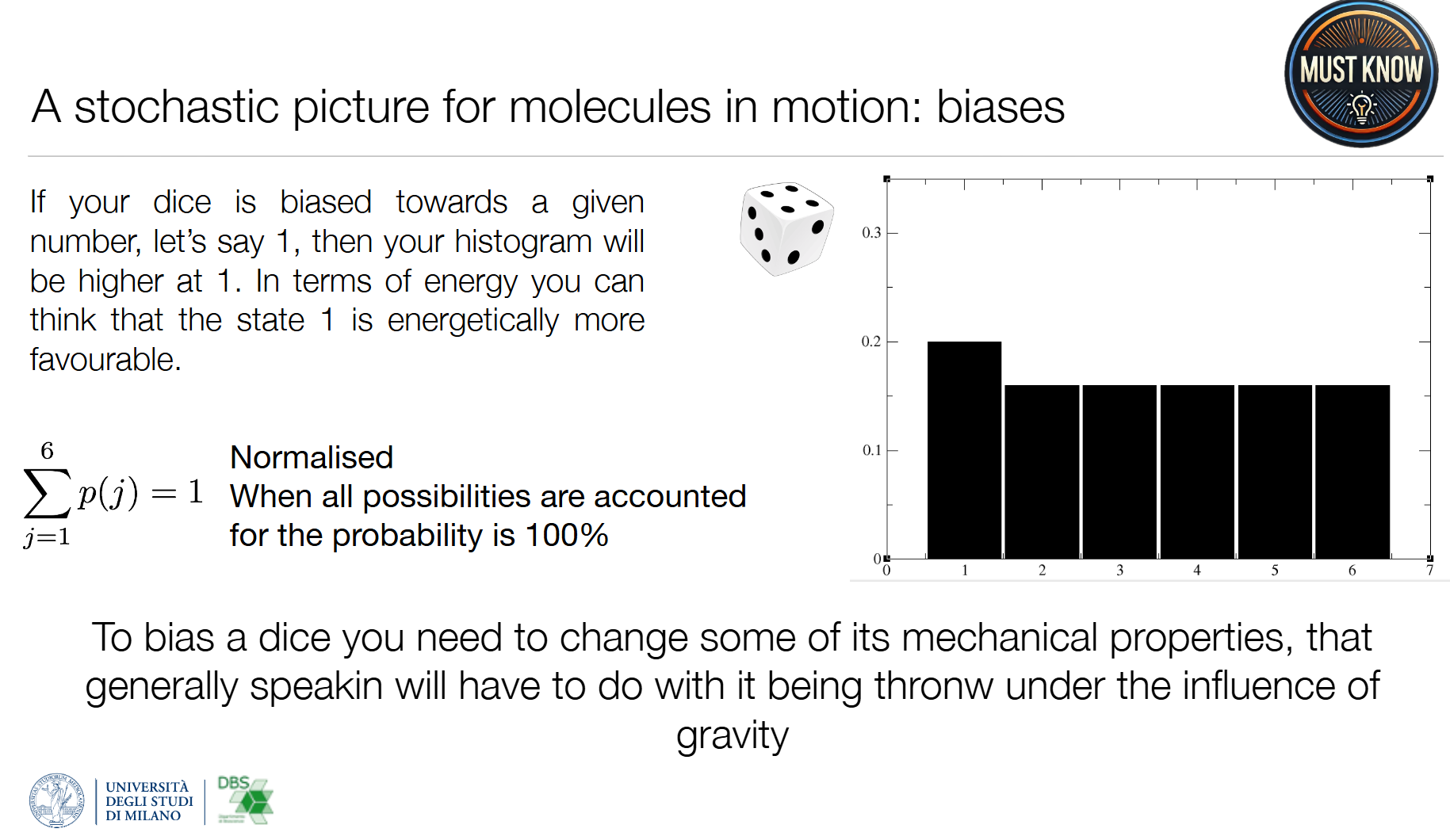
1. 离散均匀分布(公平骰子)
- 每个结果概率相等: p ( j ) = 1 / 6 p(j)=1/6 p(j)=1/6( j = 1 ∼ 6 j=1\sim6 j=1∼6)
- 归一化条件: ∑ j = 1 6 p ( j ) = 1 \sum_{j=1}^6 p(j)=1 ∑j=16p(j)=1(所有可能结果的概率总和为100%)
- 对应分子:理想无相互作用的气体分子
2. 离散有偏分布(作弊骰子)
- 某一结果概率更高,对应能量上更有利的状态
- 对应分子:蛋白质的优势构象(晶体结构就是概率最高的那个构象)
3. 连续概率分布(高斯分布)
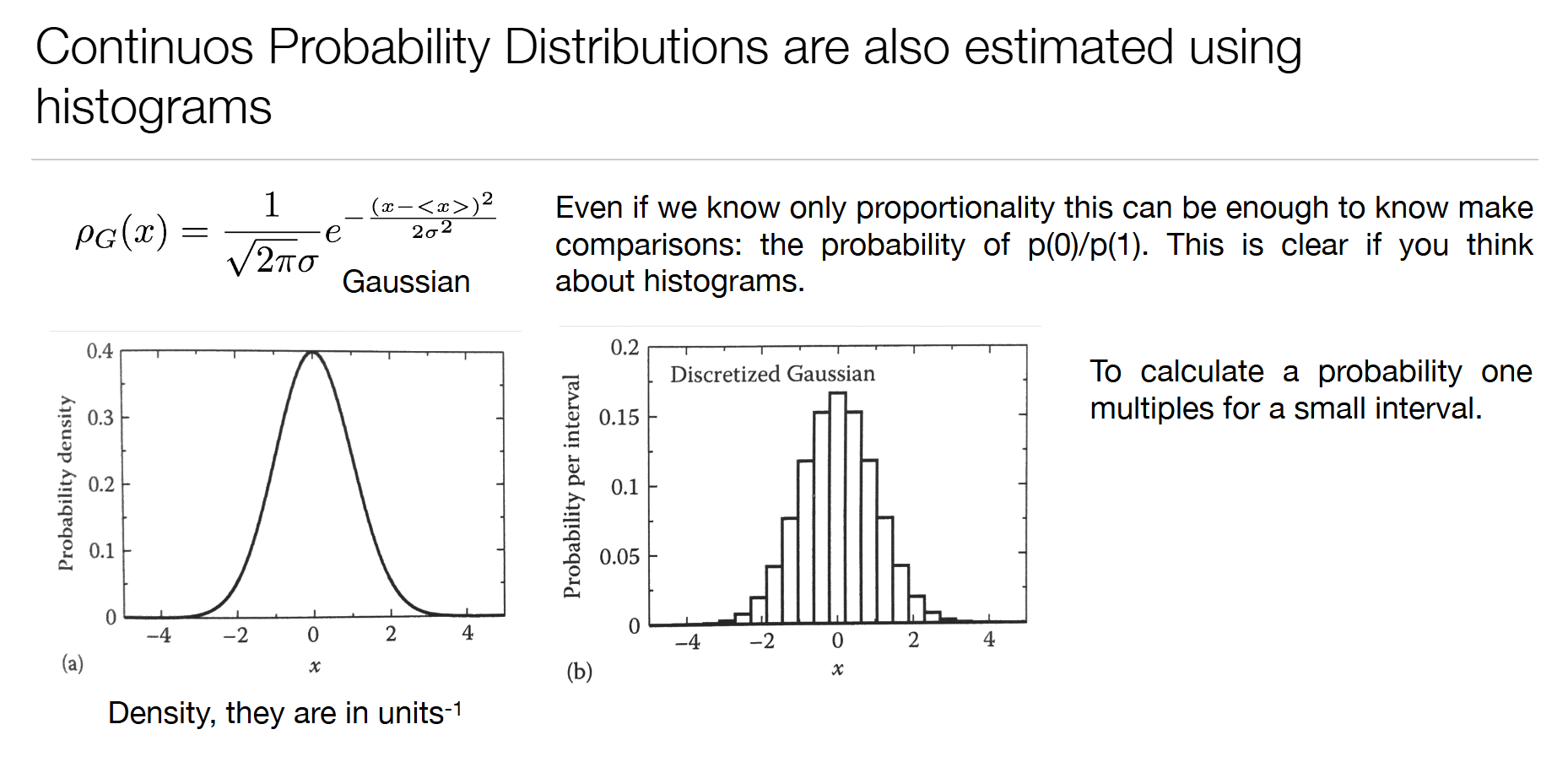
- 高斯分布公式: ρ G ( x ) = 1 2 π σ e − ( x − ⟨ x ⟩ ) 2 2 σ 2 \rho_G(x) = \frac{1}{\sqrt{2\pi}\sigma}e^{-\frac{(x-\langle x\rangle)^2}{2\sigma^2}} ρG(x)=2π σ1e−2σ2(x−⟨x⟩)2
- 连续分布用概率密度描述,概率等于密度乘以区间长度
- 对应分子:原子的热振动、分子的布朗运动
核心统计量

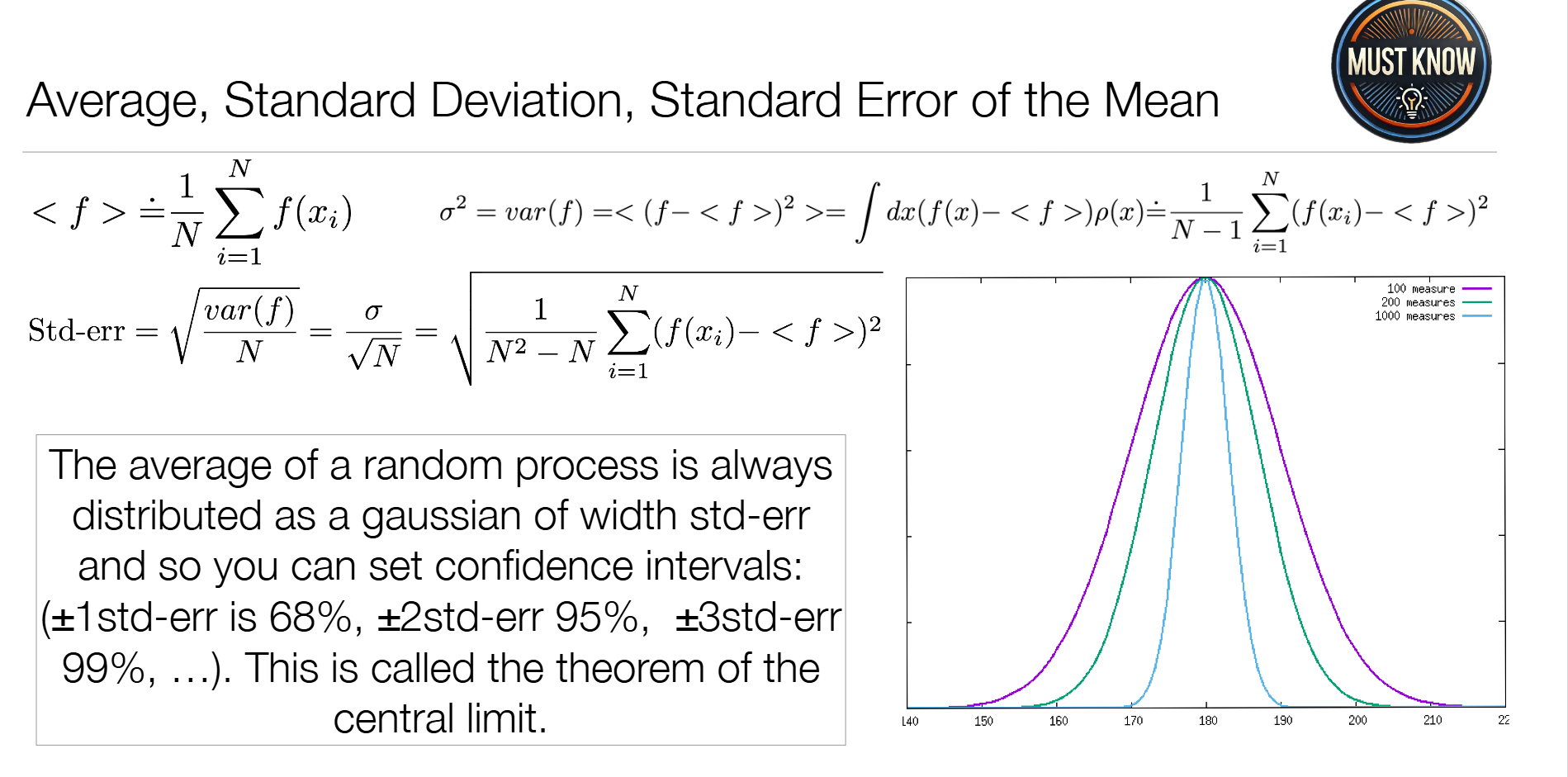
| 统计量 | 公式 | 物理意义 |
|---|---|---|
| 均值(平均值) | ⟨ f ⟩ = 1 N ∑ i = 1 N f ( x i ) \langle f\rangle = \frac{1}{N}\sum_{i=1}^N f(x_i) ⟨f⟩=N1∑i=1Nf(xi) | 分布的中心位置,对应分子的平均构象 |
| 方差/标准差 | σ 2 = 1 N − 1 ∑ i = 1 N ( f ( x i ) − ⟨ f ⟩ ) 2 \sigma^2 = \frac{1}{N-1}\sum_{i=1}^N (f(x_i)-\langle f\rangle)^2 σ2=N−11∑i=1N(f(xi)−⟨f⟩)2 | 分布的宽度,对应分子构象的波动幅度(柔性) |
| 均值标准误 | Std-err = σ N \text{Std-err} = \frac{\sigma}{\sqrt{N}} Std-err=N σ | 均值估计的准确度,采样数N越大,标准误越小,结果越可靠 |
分子动力学关键:增加模拟时间(模拟时间直接影响采样数目,增大N)可以降低标准误,提高构象平均的准确性。
多维概率分布
1. 投影(边缘分布)
- 二维分布归一化: ∫ ρ ( x , y ) d x d y = 1 \int \rho(x,y)dxdy=1 ∫ρ(x,y)dxdy=1
- 投影到x轴: ρ ( x ) = ∫ ρ ( x , y ) d y \rho(x) = \int \rho(x,y)dy ρ(x)=∫ρ(x,y)dy(忽略y变量,只看x的分布)
- 对应分子:分析蛋白质某一残基的二面角分布,忽略其他残基的影响

2. 相关性
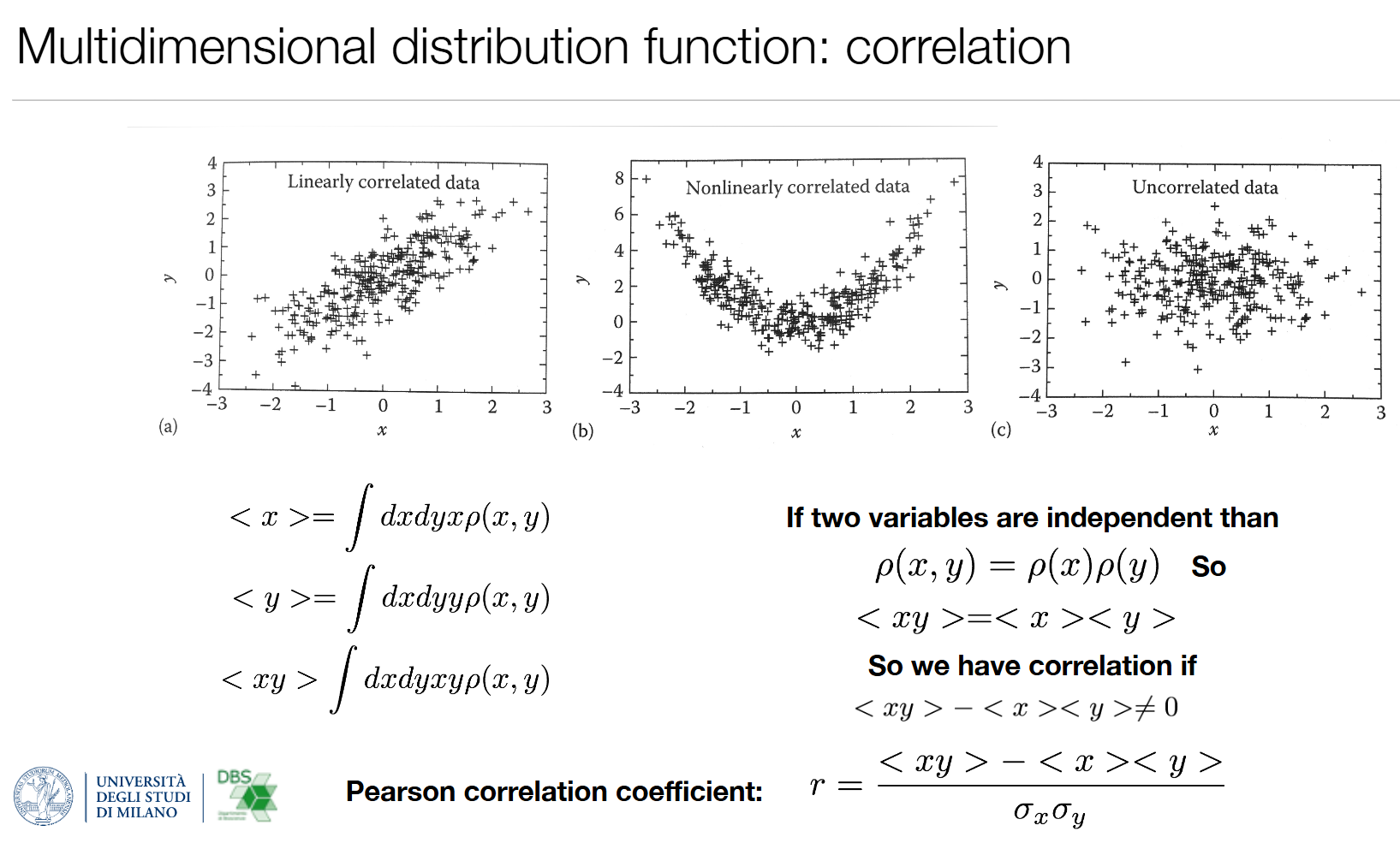
- 独立变量: ρ ( x , y ) = ρ ( x ) ρ ( y ) \rho(x,y)=\rho(x)\rho(y) ρ(x,y)=ρ(x)ρ(y),此时 ⟨ x y ⟩ = ⟨ x ⟩ ⟨ y ⟩ \langle xy\rangle=\langle x\rangle\langle y\rangle ⟨xy⟩=⟨x⟩⟨y⟩
- 相关条件: ⟨ x y ⟩ − ⟨ x ⟩ ⟨ y ⟩ ≠ 0 \langle xy\rangle - \langle x\rangle\langle y\rangle \neq 0 ⟨xy⟩−⟨x⟩⟨y⟩=0
- 皮尔逊相关系数: r = ⟨ x y ⟩ − ⟨ x ⟩ ⟨ y ⟩ σ x σ y r = \frac{\langle xy\rangle - \langle x\rangle\langle y\rangle}{\sigma_x\sigma_y} r=σxσy⟨xy⟩−⟨x⟩⟨y⟩(取值-1~1,衡量线性相关程度)
生物分子应用:分析蛋白质两个结构域的运动相关性,判断它们是否协同运动。
这里再放一点推导:



协方差这一块的话


简单来说,就是分布已知,所以确定性的积分能积出来1个确定性的值



然后再依据皮尔逊相关系数的统计定义去计算

| 图 | 数据类型 | r取值范围 | 分子力学实例 |
|---|---|---|---|
| (a) 左 | 线性正相关 | 0<r<1 | 蛋白相邻二面角协同同向扭曲 |
| (b) 中 | 非线性相关 | r≈0 | 二次势能耦合,无线性关联、但有非线性关联(⚠️ 皮尔逊只测线性,非线性相关r趋近 0) |
| © 右 | 无相关 | r≈0 | 空间相隔很远的两个残基,无相互作用 |
核心桥梁:玻尔兹曼分布
什么是玻尔兹曼分布?
在1个构象空间中,观察到某个构象的概率,用玻尔兹曼分布来描述。
我们参考维基百科:https://zh.wikipedia.org/wiki/玻尔兹曼分布
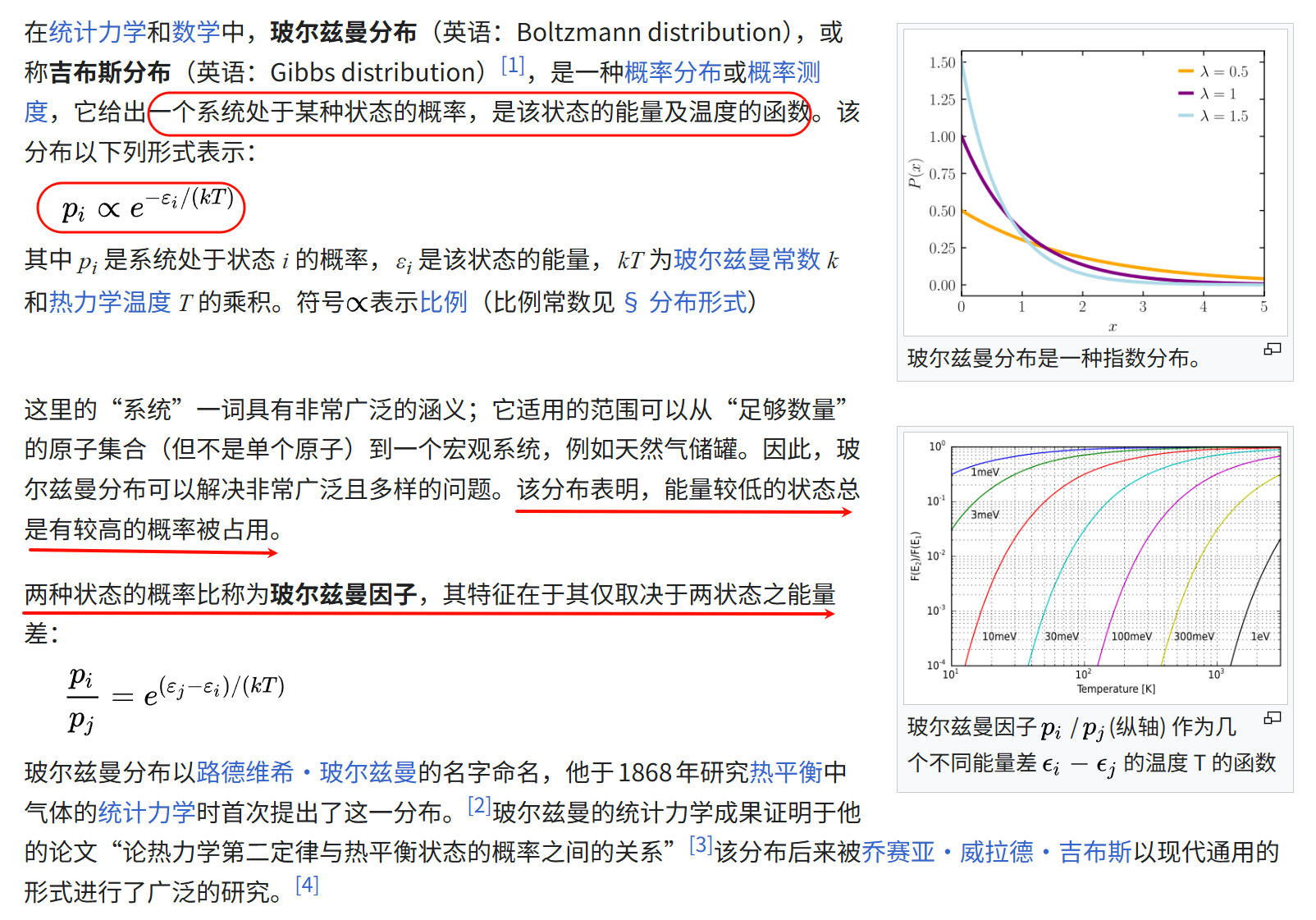
基本上只有两个概念:
- 玻尔兹曼分布:1个系统处于某种状态state i的概率,仅与其能量和温度有关(可以顺手简单写出来1个方程式)
- 玻尔兹曼因子:1个系统处于两种状态state的概率比,只与状态的能量差有关

归一化,也就是分母的系数(配分函数Z或Q),是不是看上去和softmax形式很像,
其实softmax和玻尔兹曼分布是可以联系起来的


玻尔兹曼分布与softmax的关联
本质上还是理解概率分布与能量、温度的关系。
想要深究有很多文献资料可以查阅,此处仅援引几篇高质量博客参考:
https://zhuanlan.zhihu.com/p/286724281
玻尔兹曼分布:系统处于某个能量状态i的概率 = 系统中处于能量状态i的粒子数目比例
热平衡的正则系统中,单个粒子处于能量状态 **i**的概率,等于系统内处在该能级的粒子数占总粒子数的比例。

我们先来理一下玻尔兹曼分布的推导:
- 原始的定义是从物理现象也就是实验中总结归纳出来的,也就是从分子碰撞、气体实验中总结出来的经验分布规律。这个时候就是先发现指数形式的分布结果(对应我们现成的理解的玻尔兹曼分布公式),不知道它的来源是微观状态最大化
- 后来补上了理论基石:也就是玻尔兹曼熵公式,S=kInW(熵是微观状态数的对数),这个时候才是热力学第二定律(孤立系统熵自发增大)打通了和宏观(熵)和微观(微观状态数)
- 然后我们现在理解的话,是反过来,先知道 约束+最大化微观状态------》推导分布公式,这样便于我们的理解
所以我们从理论上来推导玻尔兹曼分布公式的话,就是得先知道结果的物理意义:
我们先约束分子内能总和不变、粒子总数目不变,然后要求这个分布使得微观状态数目W最大化(最概然分布,也就是我们直接使用玻尔兹曼熵的结果,直接套用热力学第二定律的自然推导)。

那么这个时候就可以正式开始推导了,
我们的目标优化函数就是微观状态数,所以我们可以先写1个微观状态数W的函数,
假设N个粒子、K个能量状态,
也就是将N个粒子分配到K个能级,有多少种排列组合方法,这就是微观状态数W(简单记忆就是熵体现在组合上)。而这是1个经典的高中隔板排列组合问题,如何将N个小球分到K个盒子中,也就是用K-1个隔板来划分这N个球。

这里能量状态数、和微观状态数还是要区分一下。

推导如下:


- 构造微观状态数函数(目标优化函数)

- 优化形式:拉格朗日法
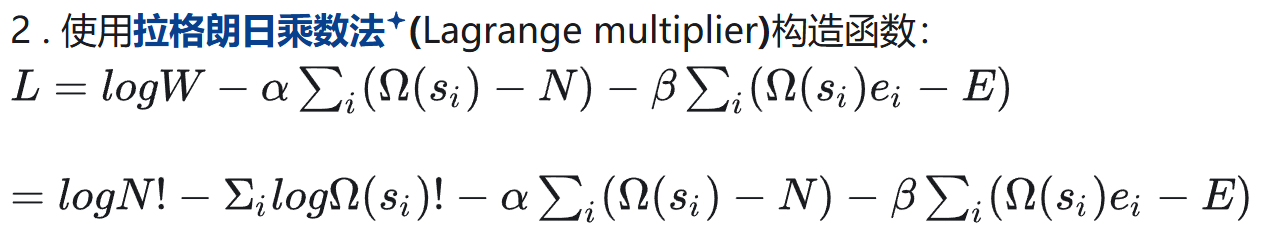
这里的目标优化函数是W,然后两个约束条件(粒子数固定、系统内容固定)

- 对拉格朗日函数的进一步简化

这里L函数的简化很简单,去掉不相关项

然后我们自变量其实有两个 引入约束项的系数、每一个微观状态数,
然后就是求偏导求极值,
我们只以其中1个自变量(微观状态数1:第1个能量级的微观状态数目)为例
形式上也很简单,就是去除不相关的变量
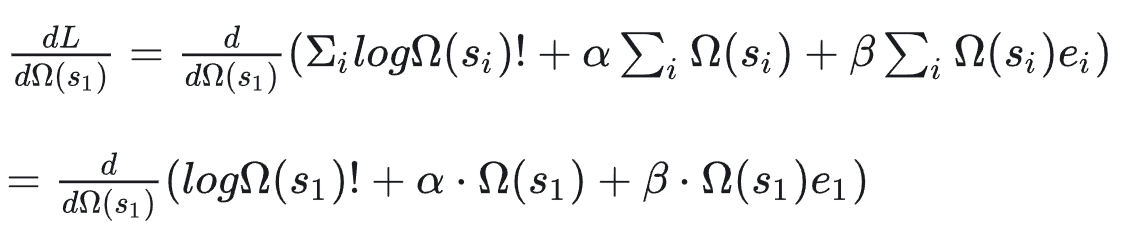

然后两个多项式求导其实挺简单的,主要是对数阶乘那个复合式子比较麻烦,
在上述展示推理中其实还是用近似(用来1个斯特林近似,就是涉及到大数阶乘n!的近似计算,因为每一个能量状态的微观粒子数目很多,基本上设计到微观体系数目量级都非常大),将其展开为多项式。
python
lnx!≈xlnx−x
x 很大时适用,统计物理求玻尔兹曼熵最常用。
然后其他的自变量同样可以求偏导数

然后分母的式子我们就也能够理解了


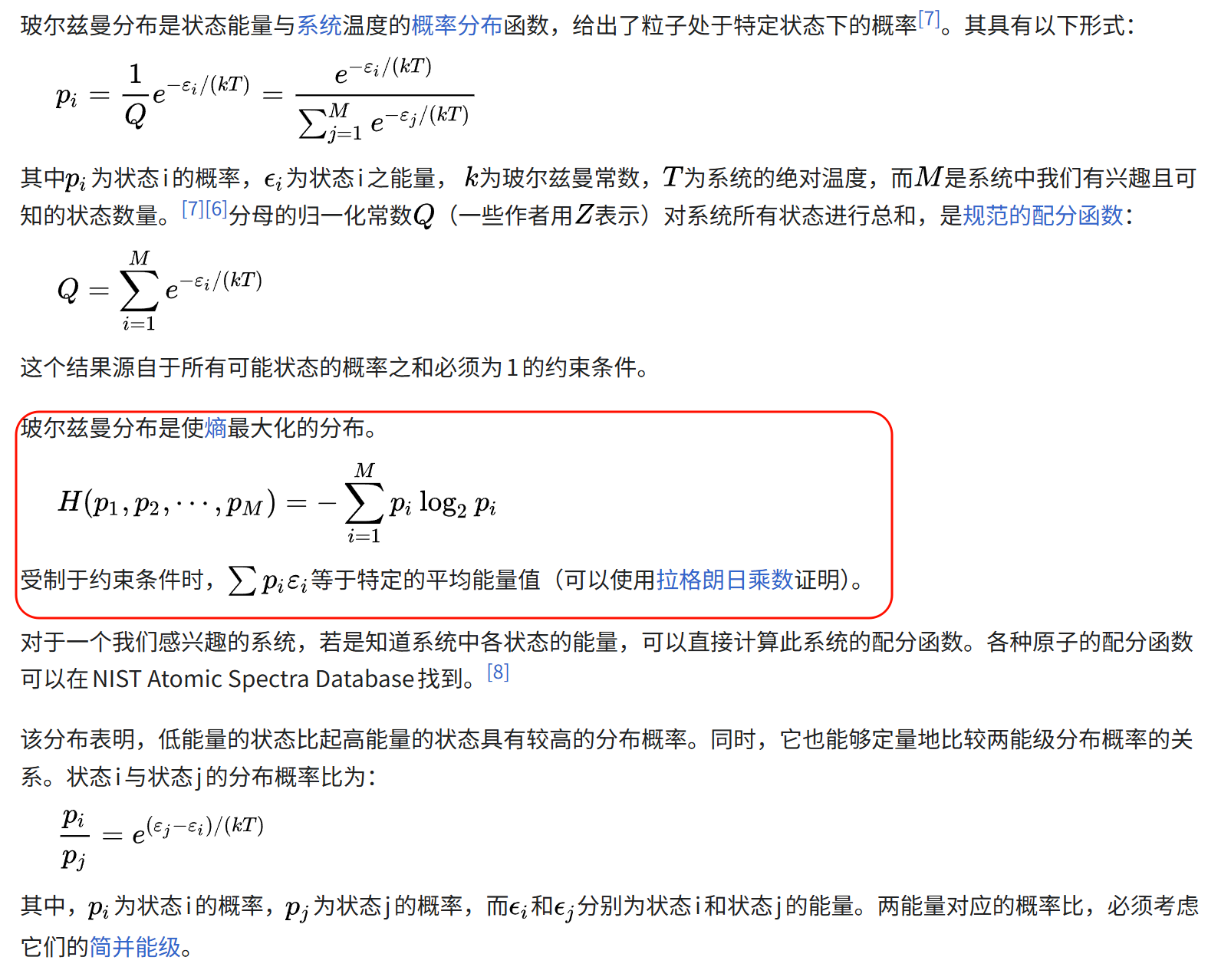
这个时候,我们再回到前面维基百科中的讨论:
玻尔兹曼分布是使熵最大化的分布,我们前面的推导就是利用了玻尔兹曼熵的公式(熵正比于微观状态数目),
所以我们用"使微观状态数最大化"的策略来推导粒子处于某一个状态的数目公式,也就是玻尔兹曼分布公式。
统计与力学的连接:玻尔兹曼分布
经过前面的简单介绍,我们基本上理解了玻尔兹曼分布的理论。

课件这里给出的公式稍微复杂了一点,就是我们考虑到一个体系处于某种状态时的概率密度函数,要从多维联合分布的角度去考虑,
比如说分子处于某一个构象,这个构象的位置、势能,和x有关;这个构象的速度、动能,和v有关。
那么这个状态的概率密度函数,其实就是x和v的联合概率密度函数,
左边的这个p我们可以拆解成多元分布,那么右边的这个e指数上的E,我们也可以将一个体系的内容拆解成势能和动能,
左边的状态概率密度函数中x、v是独立的,右边的E=势能+动能,也是解耦独立的,
然后我们再分别进行积分,就能够求得边缘分布的形式。


同理

这里稍微提一下的就是,在物理直觉上理解一下变量之间的独立

具体的推导形式要更严格一点,我们都是简单的过渡。
最终,也就是结果:
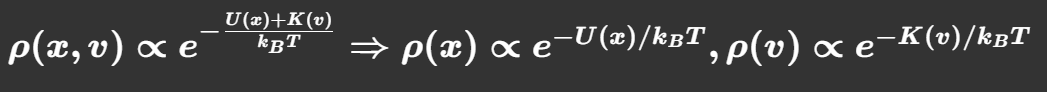
1.核心公式来源:相空间概率拆分
ρ ( x , v ) ∝ exp − E ( x , v ) k B T = exp ( − U ( x ) k B T ) ⋅ exp ( − K ( v ) k B T ) \rho(x,v)\propto \exp\left-\\frac{E(x,v)}{k_BT}\\right=\exp\left(-\frac{U(x)}{k_BT}\right)\cdot\exp\left(-\frac{K(v)}{k_BT}\right) ρ(x,v)∝exp−kBTE(x,v)=exp(−kBTU(x))⋅exp(−kBTK(v))
- E ( x , v ) = U ( x ) + K ( v ) E(x,v)=U(x)+K(v) E(x,v)=U(x)+K(v):分子总能量=构型势能 U ( x ) U(x) U(x)(位置 x x x决定,分子构象、化学键空间排布)+动能 K ( v ) = 1 2 m v 2 K(v)=\tfrac12mv^2 K(v)=21mv2**(速度** v v v决定)
- 数学上:指数乘积 = 两个独立变量的概率密度乘积 ρ ( x , v ) = ρ ( x ) ⋅ ρ ( v ) \boldsymbol{\rho(x,v)=\rho(x)\cdot\rho(v)} ρ(x,v)=ρ(x)⋅ρ(v),因此位置(构象)、速度两个随机变量统计独立
- ρ ( ⋅ ) ∝ exp ( − ... / k B T ) \rho(\cdot)\propto\exp(-\dots/k_BT) ρ(⋅)∝exp(−.../kBT):玻尔兹曼权重(Boltzmann factor) ,正比符号 ∝ \propto ∝是因为需要配分函数归一化(后面自由能公式补齐归一)
2. 拆分后两个边缘分布
- 构象分布(只关心分子空间构型,生物/大分子常用):
ρ ( x ) ∝ exp ( − U ( x ) k B T ) \boldsymbol{\rho(x)\propto \exp\left(-\frac{U(x)}{k_BT}\right)} ρ(x)∝exp(−kBTU(x))
- 忽略速度:速度积分是高斯积分 ∫ exp ( − m v 2 / 2 k B T ) d v \int \exp(-mv^2/2k_BT)dv ∫exp(−mv2/2kBT)dv是常数,归一化时被吸收,平衡构象概率只由势能 U ( x ) U(x) U(x)决定;
- 速度分布:麦克斯韦速率分布,高斯型 ρ ( v ) ∝ exp ( − 1 2 m v 2 / k B T ) \rho(v)\propto \exp\left(-\tfrac{1}{2}mv^2/k_BT\right) ρ(v)∝exp(−21mv2/kBT),热运动速度高斯分布。
注意:参考正态分布/高斯分布的基础知识, https://zh.wikipedia.org/wiki/正态分布

其实动能式子带入进去之后,我们现在的这个构象速度状态分布的概率密度,就很像正态分布/高斯分布的形式

然后高斯分布积分其实比较容易计算,这就是为什么我们一般是直接对速度变量进行边缘分布积分,因为运算好积;
如果对位置积分的话,比较难积,可能需要数值分析之类。
符号备注: k B k_B kB玻尔兹曼常数(单分子尺度,微观体系),后面出现 R R R气体常数 R = N A k B R=N_Ak_B R=NAkB(摩尔尺度,宏观热力学)是同一物理本质的尺度换算符号差异。
总而言之,玻尔兹曼分布给出了分子处于某一构象(我们容易积分v,一般只考虑x)的概率:
ρ ( x ) ∝ exp ( − U ( x ) k B T ) \rho(x) \propto \exp\left(-\frac{U(x)}{k_B T}\right) ρ(x)∝exp(−kBTU(x))
- U ( x ) U(x) U(x):构象x的势能
- k B k_B kB:玻尔兹曼常数
- T T T:绝对温度
1. 关键结论
- 能量越低,概率越高:低能构象是优势构象,这就是为什么蛋白质会折叠成特定的天然结构
- 构象与速度独立:速度分布是高斯分布,不影响构象分布,因此我们可以只关注构象的势能分布
- 温度影响分布:温度越高,高能构象的概率越大,分子的柔性越强
2. 分子动力学意义
所有分子动力学模拟的最终目的,就是按照玻尔兹曼分布采样构象空间,从而得到分子的热力学性质和动力学行为。
各态历经性(Ergodicity)/遍历性

参考:https://zh.wikipedia.org/wiki/%E9%81%8D%E6%AD%B7%E6%80%A7_(%E4%BF%A1%E8%99%9F%E8%99%95%E7%90%86)
另外参考:https://zh.wikipedia.org/wiki/遍历理论
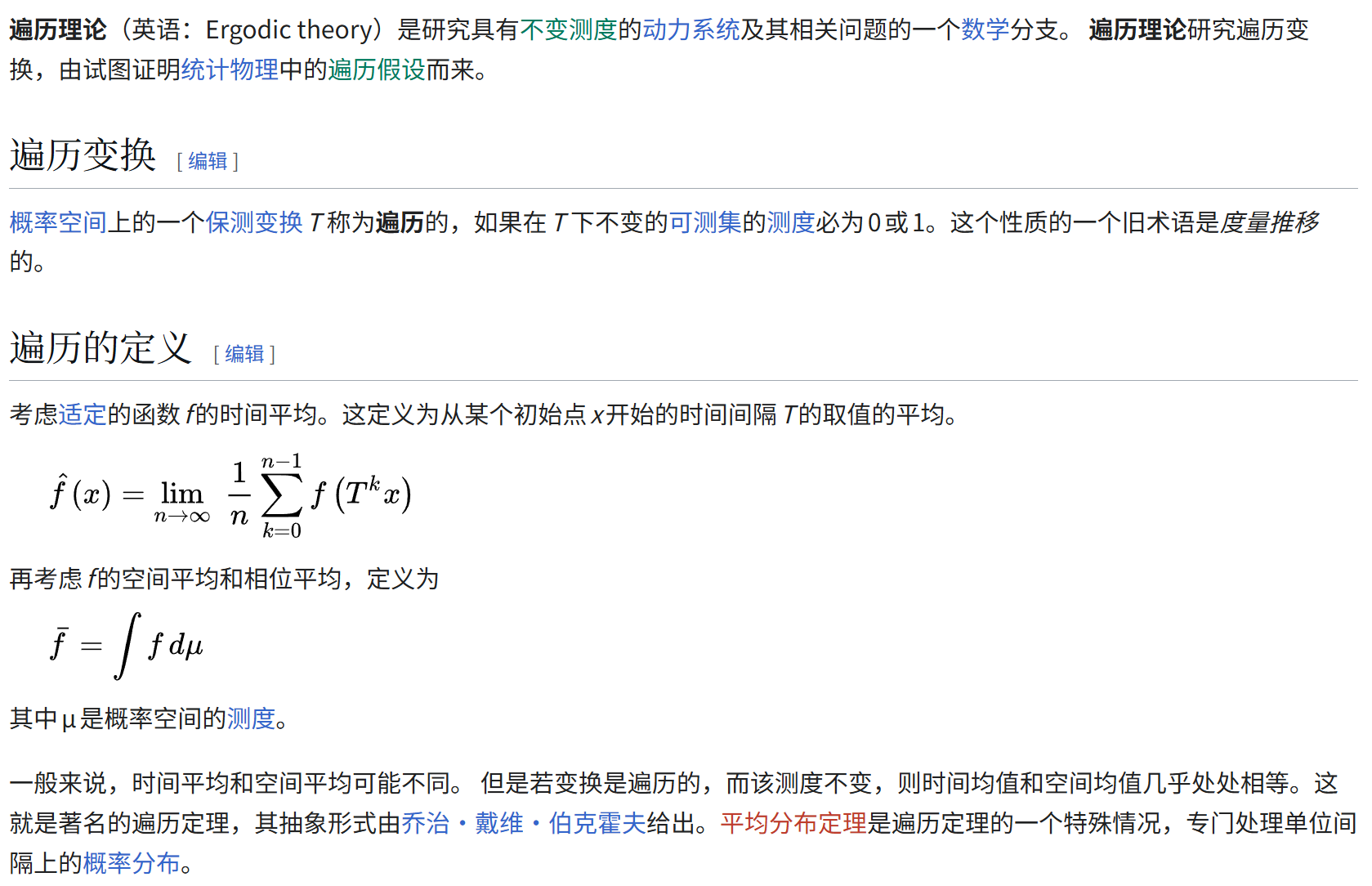
这个概念,学过随机过程,或者类似衍生应用课程(比如说通信原理)的人应该不陌生。
此处我做1个最直观的总结(仅用于理解,不一定严谨):遍历,字面意思就是每一个状态都历经过。什么叫遍历性,就是指统计结果在时间和空间上的统一性,表现为时间均值等于空间均值。
同样的,我给出1个也是非常直观的例子:
要得出一个城市A、B两座公园哪一个更受欢迎,有两种办法:
● 第一种办法:在一定的时间段考察两个公园(在空间上考察)的人数,人数多的为更受欢迎公园;
● 第二种办法:随机选择一名市民,跟踪足够长的时间(在时间上考察)来统计他去两个公园的次数,去得多的为更受欢迎公园。
一个是空间均值,一个是时间均值,如果这两个结果始终一致,则表现为遍历性。
再次借用一些经典大学物理热力学中的气体分子假想实验就是:
- 一个密闭容器,只要气体分子足够多,那么容器中的每一点都会被气体分子经过(空间平均)
- 对于单个气体分子,只要时间足够多,那么它也能够经过容器中的每一点(时间平均)
所以,物理学家一般会用一群气体分子的平均特性(空间平均),来预测单个气体分子特性(时间平均)。
也就是假设这个体系是满足遍历性的,我们用统计手段研究一群分子,就可以运用迁移到1个分子的运动规律上。
python
这个概念最早来自统计力学。 统计力学运用的是经典力学和量子力学的原理。
一个粒子运动,可以按照牛顿力学方法,计算它的运动速度、轨迹等。
但如果是大量的粒子,就很难计算,只能用统计方法计算,即概率论的方法计算。
物理学家玻尔兹曼和吉布斯假设一个密闭容器,里面有气体分子在运动,
他们不断的相互碰撞,并和容器壁碰撞,每碰撞一次,它们的运动状态就改变一次。
如果气体分子足够多,碰撞的时间足够长,那么这个密闭容器中的每一点都会被气体分子经过。
一个单独的气体分子,随着时间的流逝,也会造访容器中的每一点,
物理学家们就可以通过使用一群气体分子的平均特性,来预测单个气体分子的特性了。
所以,遍历性的学术性解释是统计结果在时间和空间上的统一性,表现为时间均值等于空间均值。总结就是,各态历经性是统计力学的核心假设,架起时间平均↔空间平均(以后我们称呼ensemble为系综,系综平均)
- 时间平均(单分子长时间演化) :单个分子在溶剂碰撞(热扰动)下,随时间随机遍历全部可能构象,在某构象停留的时间占比 = 玻尔兹曼概率ρ(x);------》恒温条件下,单个分子因与溶剂分子发生碰撞,会随机转变为所有可能的构象;各类构象的占据占比由体系能量与温度(分子和环境的相互作用)决定。
- 系综平均(瞬间快照大量全同分子):同一温度 / 体积 / 粒子数条件下,无穷多复制分子瞬间抓拍,处在某构象的分子数占比 = 同样ρ(x)。------》若在某一时刻暂停大量相同条件下的等同体系演化过程,抓拍得到的构象分布同样遵循上述统计规律。
上述两种情形下收集得到的全部构象(即取样结果)统称为构象系综(conformational ensemble,我有时候喜欢翻译为构象集合)。该系综的首要特征由取样时的环境条件决定(恒温、恒压或恒容、固定粒子数等)。
- 系综 (ensemble):满足固定T,V,N(NVT 正则系综,本 PPT 使用)的全部构象集合;由约束条件(恒温 / 恒压 / 恒粒子数)区分系综类型。
python
一、遍历性(Ergodicity):分子动力学模拟的核心假设
核心翻译与解读
时间平均:恒温下,单个分子因与溶剂碰撞会随机遍历所有可能构象,每个构象的布居度由能量和温度决定(遵循玻尔兹曼分布)。
系综平均:同一时刻,大量相同条件下的独立系统,其构象分布与单个分子长时间的构象分布完全一致。
关键结论:遍历性假设让我们可以用单个分子的长时间模拟(时间平均)代替大量分子的瞬时观测(系综平均),这是分子动力学模拟能够预测宏观性质的理论基础。
系综(Ensemble)定义
构象的集合称为系综,由其所处的热力学条件唯一确定:
NVT 系综:粒子数 (N)、体积 (V)、温度 (T) 恒定
NPT 系综:粒子数 (N)、压强 (P)、温度 (T) 恒定(最接近生物体内环境)自由能:宏观态的有效能量

自由能 F i F_i Fi定义:从微观势能 U ( x ) → U(x)\to U(x)→宏观状态有效能(微观态积分粗粒化)
1. 微观态 vs 宏观态(State)
- 微观态 :单个精确构型 x x x,能量 U ( x ) U(x) U(x);
- 宏观态A/B :同一势能阱内全部微观构型的集合(1D势能曲线两个势阱A、B),宏观态是大量微观态的打包。
2. 宏观态占据概率(积分玻尔兹曼权重)
p A = ∫ V A ρ ( x ) d x ∝ ∫ V A exp ( − U ( x ) k B T ) d x , p B = ∫ V B exp ( − U ( x ) k B T ) d x p_A=\int_{V_A}\rho(x)dx \propto \int_{V_A}\exp\left(-\frac{U(x)}{k_BT}\right)dx,\quad p_B=\int_{V_B}\exp\left(-\frac{U(x)}{k_BT}\right)dx pA=∫VAρ(x)dx∝∫VAexp(−kBTU(x))dx,pB=∫VBexp(−kBTU(x))dx
V A / V B V_A/V_B VA/VB:宏观态A/B对应的构象坐标区间(势阱定义域),宏观概率=阱内所有微观玻尔兹曼权重积分。
3. 自由能定义式(关键公式)
p A p B = ∫ V A e − U / k B T d x ∫ V B e − U / k B T d x ≡ e − F A / k B T e − F B / k B T ⟹ F i = − k B T ln ( ∫ V i exp ( − U ( x ) k B T ) d x ) \frac{p_A}{p_B}=\frac{\int_{V_A}e^{-U/k_BT}dx}{\int_{V_B}e^{-U/k_BT}dx}\equiv \frac{e^{-F_A/k_BT}}{e^{-F_B/k_BT}} \implies \boldsymbol{F_i=-k_BT\ln\left(\int_{V_i}\exp\left(-\frac{U(x)}{k_BT}\right)dx\right)} pBpA=∫VBe−U/kBTdx∫VAe−U/kBTdx≡e−FB/kBTe−FA/kBT⟹Fi=−kBTln(∫Viexp(−kBTU(x))dx)
指数上的这个空间积分比较难算,所以直接把这些构象空间Va、Vb庞大的积分结果打包成1个宏观的热力学量,也就是自由能Fi

- 物理意义:自由能 F i F_i Fi是宏观态的等效能量 :不用逐个算无穷多微观 U ( x ) U(x) U(x),只用一个等效 F i F_i Fi就能用玻尔兹曼公式直接写宏观态概率;
- 对数+积分项本质:积分 ∫ e − U / k B T d x = Z i \int e^{-U/k_BT}dx=Z_i ∫e−U/kBTdx=Zi是子配分函数 , F = − k B T ln Z F=-k_BT\ln Z F=−kBTlnZ是正则系综亥姆霍兹自由能标准定义;
- 熵隐藏在积分里:积分越大→同一势阱内微观构象数目越多(简并度大,熵高)→ F F F越小(自由能=内能-TS,熵大自由能更低)。
1. 状态(State)的定义
状态是属于同一势能谷的所有微观构象的集合。例如,蛋白质的"折叠态"和"去折叠态"就是两个不同的宏观态,每个态包含天文数字级别的微观构象。
2. 自由能与概率的关系
对于一维势能面中的两个状态A和B:
- 状态A的概率: p A = ∫ V A ρ ( x ) d x ∝ ∫ V A exp ( − U ( x ) k B T ) d x p_A = \int_{V_A} \rho(x)dx \propto \int_{V_A} \exp\left(-\frac{U(x)}{k_B T}\right)dx pA=∫VAρ(x)dx∝∫VAexp(−kBTU(x))dx
- 状态B的概率: p B = ∫ V B ρ ( x ) d x ∝ ∫ V B exp ( − U ( x ) k B T ) d x p_B = \int_{V_B} \rho(x)dx \propto \int_{V_B} \exp\left(-\frac{U(x)}{k_B T}\right)dx pB=∫VBρ(x)dx∝∫VBexp(−kBTU(x))dx
自由能是宏观态的"有效能量",它将一个态的所有微观构象的能量整合为一个单一数值:
p A p B = exp ( − F A k B T ) exp ( − F B k B T ) \frac{p_A}{p_B} = \frac{\exp\left(-\frac{F_A}{k_B T}\right)}{\exp\left(-\frac{F_B}{k_B T}\right)} pBpA=exp(−kBTFB)exp(−kBTFA)
F i = − k B T ln ( ∫ V i exp ( − U ( x ) k B T ) d x ) F_i = -k_B T \ln\left(\int_{V_i} \exp\left(-\frac{U(x)}{k_B T}\right)dx\right) Fi=−kBTln(∫Viexp(−kBTU(x))dx)
核心区别:
- 势能 U ( x ) U(x) U(x):单个微观构象的能量
- 自由能 F i F_i Fi:整个宏观态的有效能量,包含了熵的贡献(即该态包含的微观构象数目)
能量 - 概率 - 时间的统一关系

1. 微观与宏观的分野
| 尺度 | 主导量 | 关系 |
|---|---|---|
| 微观过程 | 势能U | 两个微观态的相对概率与它们的势能差呈指数关系 |
| 宏观过程 | 自由能F | 两个宏观态的相对概率与它们的自由能差呈指数关系 |
2. 时间尺度与自由能垒
宏观过程的平均发生时间由分隔两个态的自由能垒高度决定:能垒越高,过程越慢。这就是为什么有些蛋白质折叠需要微秒级,而有些需要毫秒甚至秒级。
微观规律↔宏观规律总结(能量 U U U vs 自由能 F F F;概率指数依赖能量差)
- 微观过程(单构型间跃迁) :两个微观态 x 1 , x 2 x_1,x_2 x1,x2,相对概率 p ( x 1 ) p ( x 2 ) ∝ exp ( − U 1 − U 2 k B T ) \displaystyle \frac{p(x_1)}{p(x_2)}\propto\exp\left(-\frac{U_1-U_2}{k_BT}\right) p(x2)p(x1)∝exp(−kBTU1−U2),由势能差 Δ U ΔU ΔU控制;
- 宏观过程(A/B宏观态跃迁) :两个宏观态, p A p B ∝ exp ( − F A − F B k B T ) \displaystyle \frac{p_A}{p_B}\propto\exp\left(-\frac{F_A-F_B}{k_BT}\right) pBpA∝exp(−kBTFA−FB),由自由能差 Δ F ΔF ΔF控制;
宏观态包含大量微观态,熵(微观态数目)被整合进自由能 F F F,所以宏观不再用原始势能 U U U,改用粗粒化自由能 F F F;
- 动力学:跃迁时间和自由能势垒 :宏观转变的平均等待时间 τ ∝ exp ( F ‡ − F state k B T ) \tau\propto\exp\left(\frac{F^\ddagger-F_\text{state}}{k_BT}\right) τ∝exp(kBTF‡−Fstate)(阿伦尼乌斯规律),势垒 F ‡ F^\ddagger F‡越高,跨越越慢、时间越长。
溶液中分子的随机动力学图像
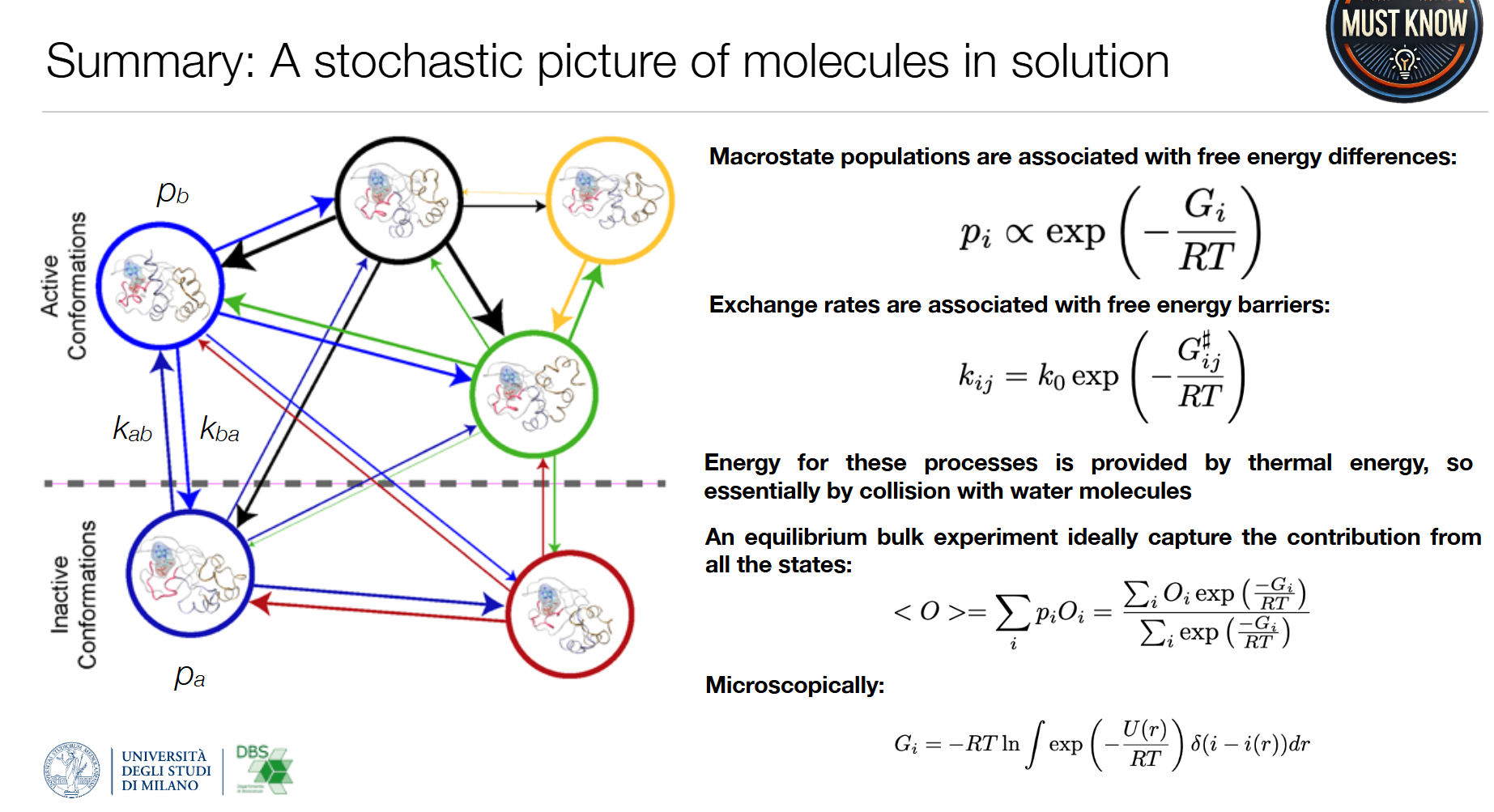
- 宏观态布居 : p i ∝ exp ( − G i R T ) p_i \propto \exp\left(-\frac{G_i}{RT}\right) pi∝exp(−RTGi)( G 为吉布斯自由能, 为吉布斯自由能, 为吉布斯自由能,R为气体常数)
- 态间交换速率 : k i j = k 0 exp ( − G i j ‡ R T ) k_{ij} = k_0 \exp\left(-\frac{G_{ij}^\ddagger}{RT}\right) kij=k0exp(−RTGij‡)(阿伦尼乌斯方程, G i j ‡ G_{ij}^\ddagger Gij‡为从态i到态j的自由能垒)
- 平均观测值:平衡态实验测量的是所有态的加权平均
⟨ O ⟩ = ∑ i p i O i = ∑ i O i exp ( − G i R T ) ∑ i exp ( − G i R T ) \langle O \rangle = \sum_i p_i O_i = \frac{\sum_i O_i \exp\left(-\frac{G_i}{RT}\right)}{\sum_i \exp\left(-\frac{G_i}{RT}\right)} ⟨O⟩=∑ipiOi=∑iexp(−RTGi)∑iOiexp(−RTGi)
所有这些过程的能量来源都是热能,即分子与水分子的随机碰撞。
生物大分子随机模型(符号从 k B T → R T k_BT\to RT kBT→RT; F → G F\to G F→G吉布斯自由能,摩尔热力学)
符号变更说明(最关键差异:单分子 k B k_B kB ↔ 摩尔量 R R R; F F F亥姆霍兹→ G G G吉布斯)
- R T = N A k B T RT=N_Ak_BT RT=NAkBT: R R R气体常数(1mol分子尺度,生化常用摩尔热力学), k B k_B kB单分子玻尔常数;
- G i G_i Gi:吉布斯自由能(恒压条件 N P T NPT NPT**,溶液生化实验恒压)** ,前面 F F F是恒容亥姆霍兹自由能;实验溶液大多常压,因此换成 G G G,公式形式完全不变,只是常数替换: F = − k B T ln Z → G = − R T ln Z F=-k_BT\ln Z \to G=-RT\ln Z F=−kBTlnZ→G=−RTlnZ。
3组核心公式
- 平衡布居概率(宏观态占比)
p i ∝ exp ( − G i R T ) \boldsymbol{p_i\propto \exp\left(-\frac{G_i}{RT}\right)} pi∝exp(−RTGi)
- 多个构象宏观态(激活/失活等不同蛋白构象环),平衡时丰度由各自吉布斯自由能决定, G i G_i Gi越低, p i p_i pi越大;
- 态间跃迁速率(阿伦尼乌斯/过渡态理论)
k i j = k 0 exp ( − G i j ‡ R T ) \boldsymbol{k_{ij}=k_0\exp\left(-\frac{G^\ddagger_{ij}}{RT}\right)} kij=k0exp(−RTGij‡)
- G i j ‡ G^\ddagger_{ij} Gij‡: i → j i\to j i→j跨越的过渡态自由能势垒;势垒越高,速率常数 k i j k_{ij} kij越小、转变越慢; k 0 k_0 k0是前置因子(碰撞频率,溶剂水分子热碰撞提供翻越势垒的能量);
- 实验可观测量系综平均
⟨ O ⟩ = ∑ i p i O i = ∑ i O i exp ( − G i / R T ) ∑ i exp ( − G i / R T ) \langle O\rangle=\sum_i p_i O_i=\frac{\sum_i O_i \exp(-G_i/RT)}{\sum_i \exp(-G_i/RT)} ⟨O⟩=∑ipiOi=∑iexp(−Gi/RT)∑iOiexp(−Gi/RT)
- 宏观实验测到的信号 ⟨ O ⟩ \langle O\rangle ⟨O⟩是所有构象态 i i i的观测值 O i O_i Oi按玻尔兹曼概率加权平均(比如NMR、荧光测蛋白平均信号,是全部构象加权结果);
- 微观定义回代: G i = − R T ln ∫ exp ( − U ( r ) R T ) δ ( i − i ( r ) ) d r \displaystyle G_i=-RT\ln\int \exp\left(-\frac{U(r)}{RT}\right)\delta(i-i(r))dr Gi=−RTln∫exp(−RTU(r))δ(i−i(r))dr
δ \delta δ函数筛选所有属于宏观态 i i i的微观坐标 r r r,积分就是该态配分函数,和第3页 F i = − k B T ln ∫ e − U / k B T d x F_i=-k_BT\ln\int e^{-U/k_BT}dx Fi=−kBTln∫e−U/kBTdx数学结构完全一致,仅 k B T ↔ R T 、 F ↔ G k_BT\leftrightarrow RT、F\leftrightarrow G kBT↔RT、F↔G尺度/热力学条件替换。
符号差异汇总
| 符号 | 物理含义 | 使用场景 |
|---|---|---|
| k B k_B kB玻尔兹曼常数 | 单分子微观能量-温度关联 | 理论微观势能 U U U、亥姆霍兹自由能 F F F(前3页) |
| R R R气体常数 | 1摩尔分子能量-温度关联 | 生化宏观实验、吉布斯自由能 G G G(最后一页) |
| U ( x ) U(x) U(x) | 微观单个构型势能 | 单个分子空间构象的势能(最底层微观能量) |
| F i F_i Fi | 亥姆霍兹自由能(NVT恒容) | 理论模型,恒容体系宏观态等效能 |
| G i G_i Gi | 吉布斯自由能(NPT恒压) | 溶液生物实验(常压),实际测量体系 |
| ρ ( x ) \rho(x) ρ(x) | 微观构象概率密度 | 单分子连续坐标分布 |
| p i p_i pi | 宏观态离散占据概率 | 打包后的势能阱宏观态布居 |
整体理论逻辑链
微观势能 U ( x ) → 玻尔兹曼权重 exp ( − U / k B T ) 微观构象分布 ρ ( x ) → 势阱内积分 ∫ d x 宏观态配分函数 → − k B T ln ( ⋅ ) 自由能定义 F / G → exp ( − G / R T ) 宏观态概率 p i → 势垒 跃迁速率 k i j \boldsymbol{微观势能U(x)\xrightarrow\\text{玻尔兹曼权重}{\exp(-U/k_BT)}微观构象分布\rho(x)\xrightarrow\\text{势阱内积分}{\int dx}宏观态配分函数\xrightarrow-k_BT\\ln(\\cdot){\text{自由能定义}}F/G \xrightarrow{\exp(-G/RT)}宏观态概率p_i \xrightarrow{\text{势垒}}跃迁速率k_{ij}} 微观势能U(x)exp(−U/kBT) 玻尔兹曼权重微观构象分布ρ(x)∫dx 势阱内积分宏观态配分函数自由能定义 −kBTln(⋅)F/Gexp(−G/RT) 宏观态概率pi势垒 跃迁速率kij
热运动(溶剂分子碰撞)提供能量,让分子在不同宏观构象间随机跃迁;平衡时各构象丰度由自由能决定、跃迁快慢由自由能势垒决定,全部宏观热力学行为根源都是微观势能+玻尔兹曼统计+热扰动。
生物分子过程实例
配体-受体结合

1. 反应与速率常数定义
R + L ⇌ k o n k o f f R L \boldsymbol{R+L \underset{k_{off}}{\stackrel{k_{on}}{\rightleftharpoons}} RL} R+Lkoff⇌konRL
| 参数 | 物理含义 | 单位 | 微观本质 |
|---|---|---|---|
| k o n k_{on} kon(结合速率) | 二级结合速率, k o n R L k_{on}RL konRL=每秒生成RL复合物数目 | M − 1 ⋅ s − 1 \mathrm{M^{-1}\cdot s^{-1}} M−1⋅s−1 | ①配体与受体溶液碰撞;②碰撞后形成稳定复合物, k o n = k_{on}= kon=碰撞频率×结合成功概率 |
| k o f f k_{off} koff(解离速率) | 一级解离速率, k o f f R L k_{off}RL koffRL=每秒RL解离为R+L的数目 | s − 1 \mathrm{s^{-1}} s−1 | 复合物受热扰动、打破分子间相互作用,从结合态变为游离态的频率 |
过量配体条件 L ≫ R L\ggR L≫R时, k o n L k_{on}L konL等效为伪一级结合速率( s − 1 \mathrm{s^{-1}} s−1)。
2. 平衡条件与解离常数 K d K_d Kd推导
平衡时:单位时间结合总数=解离总数
k o n R L = k o f f R L k_{on}RL=k_{off}RL konRL=koffRL
变形得到解离常数:
K d = k o f f k o n = R L R L \boldsymbol{K_d=\frac{k_{off}}{k_{on}}=\frac{RL}{RL}} Kd=konkoff=RLRL
- C 0 = 1 M C^0=1\ \mathrm{M} C0=1 M为热力学标准浓度;
- 概率关联: K d = C 0 ⋅ p u n b o u n d p b o u n d \displaystyle K_d=C^0\cdot\frac{p_{\mathrm{unbound}}}{p_{\mathrm{bound}}} Kd=C0⋅pboundpunbound
p u n b o u n d p_{\mathrm{unbound}} punbound:受体游离概率; p b o u n d p_{\mathrm{bound}} pbound:受体被配体结合概率。 - 规律: K d \boldsymbol{K_d} Kd数值越小→ p b o u n d p u n b o u n d \displaystyle\frac{p_{\mathrm{bound}}}{p_{\mathrm{unbound}}} punboundpbound越大→结合亲和力越强。
3. 结合自由能(热力学-概率桥梁,承接玻尔兹曼)
Δ G b i n d i n g = k B T ln K d C 0 = k B T ln ( p u n b o u n d p b o u n d ) \boldsymbol{\Delta G_{\mathrm{binding}}=k_BT\ln\frac{K_d}{C^0}=k_BT\ln\left(\frac{p_{\mathrm{unbound}}}{p_{\mathrm{bound}}}\right)} ΔGbinding=kBTlnC0Kd=kBTln(pboundpunbound)
- Δ G < 0 \Delta G<0 ΔG<0:结合自发,结合态自由能更低、占据概率更高(玻尔兹曼分布 p ∝ e − G / k B T p\propto e^{-G/k_BT} p∝e−G/kBT);
- 物理意义:结合自由能是把游离R+L变为结合RL所需的热力学功,直接关联宏观态概率比值。
蛋白质折叠
1. 二态折叠反应
U ⇌ k f k u F \boldsymbol{U \underset{k_u}{\stackrel{k_f}{\rightleftharpoons}} F} Uku⇌kfF
U U U=去折叠无序态; F F F=天然折叠有序态
- k f k_f kf:折叠速率( s − 1 \mathrm{s^{-1}} s−1), k f P k_fP kfP=每秒折叠成天然态分子数;
- k u k_u ku:去折叠速率( s − 1 \mathrm{s^{-1}} s−1), k u P k_uP kuP=每秒天然态解折叠数目。
2. 平衡常数与折叠自由能
平衡: k f U = k u F k_fU=k_uF kfU=kuF
K D = k u k f , Δ G e q = R T ln K D K_D=\frac{k_u}{k_f},\quad \boldsymbol{\Delta G_{eq}=RT\ln K_D} KD=kfku,ΔGeq=RTlnKD
Δ G e q < 0 \Delta G_{eq}<0 ΔGeq<0:天然折叠态 F F F自由能更低,平衡下天然态占比更高(概率更大)。
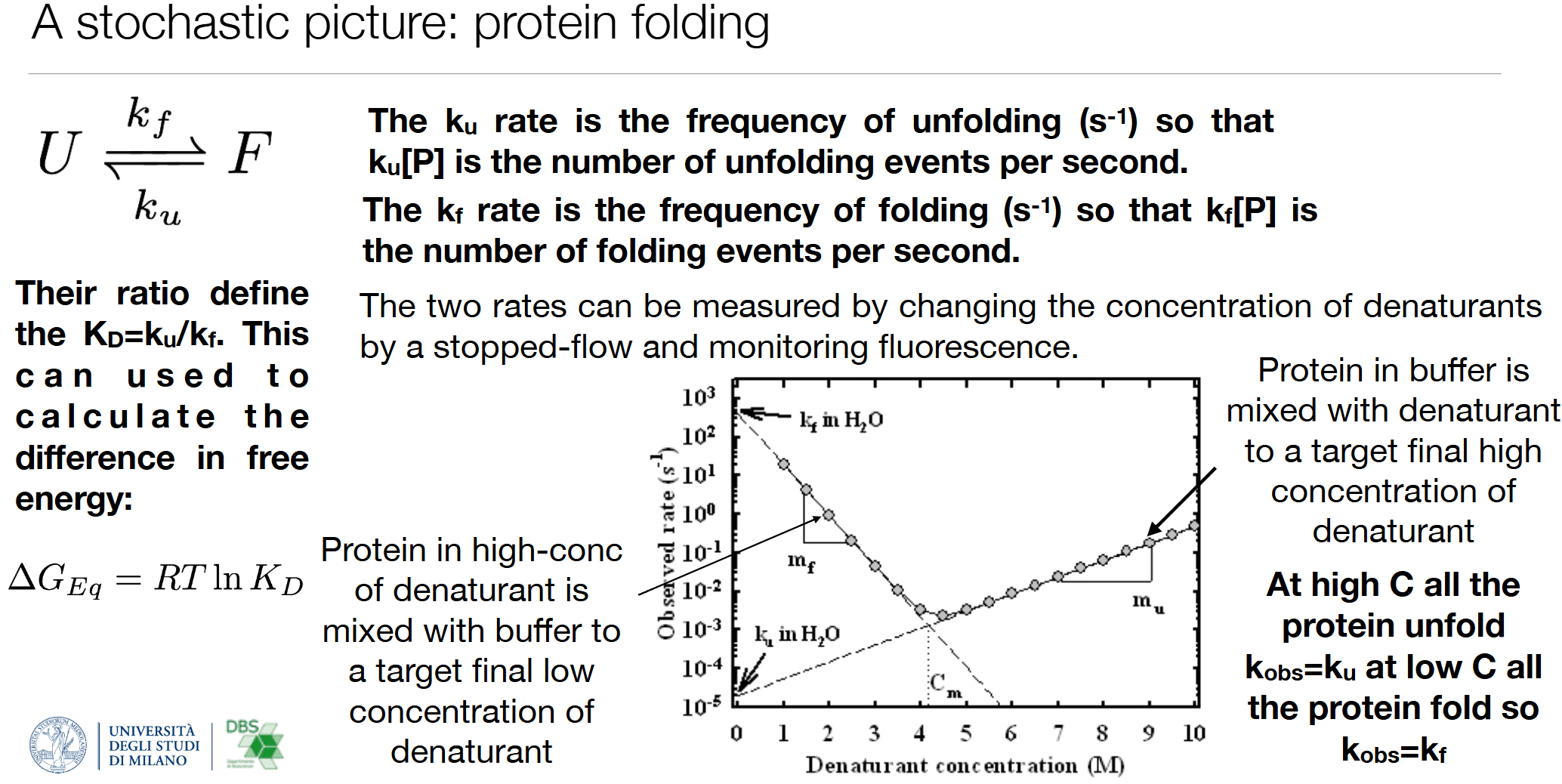
3. 实验测定方法(变性剂梯度+停流荧光Stopped-flow)
- 高浓度变性剂:蛋白全部去折叠,表观速率 k o b s = k u k_{\mathrm{obs}}=k_u kobs=ku;
- 低浓度变性剂:蛋白全部折叠,表观速率 k o b s = k f k_{\mathrm{obs}}=k_f kobs=kf;
- 改变变性剂浓度做速率曲线,外推至纯水条件,得到生理条件 k f 、 k u k_f、k_u kf、ku,进而算折叠自由能 Δ G e q \Delta G_{eq} ΔGeq;
C m C_m Cm:折叠-去折叠各占50%的变性剂临界浓度。
结构生物信息学统一纲领(收尾逻辑)
两大永恒核心问题:采样(Sampling) + 能量(Energy/打分Score/损失Loss)

- 能量/打分函数 :等效统计力学里的势能 U U U,用来定量判断构象优劣(能量越低、构象热力学概率越高,对应AI里损失越小、模型越优);
- 采样:在庞大构象空间里,按照玻尔兹曼概率规则取样,对应MD/MC模拟采样、AI数据集采样;
全领域通用:
MD分子动力学、分子对接(Docking)、蛋白结构预测、蛋白质设计、机器学习/AI蛋白模型,全部围绕「怎么高效采样、怎么精准定义能量/打分」两个问题。
统计和力学交汇的地方就在于,我们能够用力学的能量概念来解释概率分布观测到的现象,
力学中的能量,就是概率中的score、loss函数。
一、底层物理逻辑:玻尔兹曼分布是二者的数学桥梁
- 经典力学维度
能量 E ( r ) E(\boldsymbol r) E(r)由分子内化学键、范德华、静电等相互作用唯一确定,是描述单个微观构象稳定性的力学标尺,只遵从受力、势能转化的力学规律。 - 统计学维度
平衡体系中构象出现概率严格遵循玻尔兹曼规律:
p ( r ) ∝ exp ( − E ( r ) k B T ) \boldsymbol{p(\boldsymbol r)\propto \exp\left(-\frac{E(\boldsymbol r)}{k_B T}\right)} p(r)∝exp(−kBTE(r))
- 能量越低,构象在采样、实验观测里出现的统计概率越高。
我们在MD模拟、荧光实验里统计得到的构象频次分布(观测概率),全部可以反向用能量高低做解释,实现「力学量→统计观测现象」的定量闭环,这就是两门学科的交汇本质。
二、一一对应:物理能量 ↔ 打分Score / 损失Loss(结构生信&AI通用等价关系)
1. 分子对接、传统结构预测:势能=Score打分函数
- 物理能量:原子层面真实势能 U ( r ) U(\boldsymbol r) U(r);
- 工程化Score:简化后的经验/半经验势能,打分数值越小=等效能量越低=构象热力学概率越高;
- 对接筛选逻辑:优先保留高分(低能)构象,本质就是按玻尔兹曼概率择优采样。
2. 蛋白质AI结构预测:自由能=Loss损失函数
深度学习训练最小化Loss,等价于热力学体系趋向最小自由能;
模型生成构象的概率分布满足: p ∝ e − L o s s / T p\propto e^{-\mathrm{Loss}/T} p∝e−Loss/T,Loss越小,模型输出该构象的概率越大;
本质:Score、Loss都是物理能量的抽象变形,舍弃了原子势能的精细物理参数,但完整继承了「数值越低,状态统计占比越高」的统计力学核心规则。
所有结构生物信息学方法,包括:
- 分子动力学模拟(MD)
- 蒙特卡洛模拟(MC)
- 分子对接
- 蛋白质结构预测
- 蛋白质设计
都面临两个核心问题:
- 采样问题:如何高效探索巨大的构象空间
- 能量问题:如何准确描述构象的能量(即打分函数/损失函数)
后续课程将围绕这两个问题,介绍不同的解决策略和算法。
- 微观单构象:势能 U → 玻尔兹曼 p ∝ e − U / k B T U\xrightarrow{\mathrm{玻尔兹曼}}p\propto e^{-U/k_BT} U玻尔兹曼 p∝e−U/kBT(单点构象概率)
- 宏观多构象态:自由能 F → 自由能公式 p m a c r o ∝ e − F / k B T F\xrightarrow{\mathrm{自由能公式}}p_{\mathrm{macro}}\propto e^{-F/k_BT} F自由能公式 pmacro∝e−F/kBT(折叠/结合宏观态概率)
- 动力学层面:速率 k ∝ e − G ‡ / k B T k\propto e^{-G^\ddagger/k_BT} k∝e−G‡/kBT(能垒决定反应快慢, k o f f / k o n 、 k u / k f k_{off}/k_{on}、k_u/k_f koff/kon、ku/kf)
- 实验/计算:通过 k k k测 K d / K D K_d/K_D Kd/KD→反推自由能 Δ G \Delta G ΔG,用MD/AI采样验证概率分布。
如何从统计物理的角度来理解深度学习?
这其实是一个非常艰深的领域,也是一个非常重要、复杂的问题。
前面我们关于玻尔兹曼分布和softmax联系的简单讨论其实已经有一点点韵味在里面了,
总而言之,物理直觉、物理类比,以及很多物理可解释性,将会越来越重要。
这里推荐一篇很棒的总结性推文:https://github.com/datawhalechina/Path2AGI/blob/main/24-statistical-physics.md
对关于统计物理和AI的理论关联感兴趣的强烈建议去阅读一下,对于AI的底层理解能够拔高1个层次!
建议仅作为深入研学的起点,相关学习依然需要系统性阅读文献。




主要是介绍了统计物理对 AI/AGI 发展的核心贡献,提供了理解分布、能量、熵、自由能和不可逆过程的统一语言,为能量模型、变分推断、扩散模型等方法提供了理论启发。
Notebooks 练习
课堂练习

- 如何计算随机过程的平均结果?
对大量独立观测值取算术平均: ⟨ f ⟩ = 1 N ∑ i = 1 N f ( x i ) \langle f\rangle = \frac{1}{N}\sum_{i=1}^N f(x_i) ⟨f⟩=N1∑i=1Nf(xi) - 标准差和均值标准误的区别是什么?
- 标准差 σ \sigma σ:描述分布本身的宽度,反映数据的离散程度
- 均值标准误 Std-err = σ / N \text{Std-err} = \sigma/\sqrt{N} Std-err=σ/N :描述均值估计的准确度,反映多次测量均值的离散程度
- 过程平均值的概率分布是什么?
根据中心极限定理,无论原分布是什么,平均值的分布都趋近于高斯分布。 - 事件的绝对概率和相对概率的区别是什么?
- 绝对概率:事件发生的真实概率,总和为1
- 相对概率:两个事件发生概率的比值,不需要知道所有可能结果
- 宏观态的概率与其自由能的关系是什么?
宏观态的概率与自由能呈指数关系: p i ∝ exp ( − G i R T ) p_i \propto \exp\left(-\frac{G_i}{RT}\right) pi∝exp(−RTGi),自由能越低,概率越高。 - 构象的概率与其能量的关系是什么?
微观构象的概率与势能呈指数关系: ρ ( x ) ∝ exp ( − U ( x ) k B T ) \rho(x) \propto \exp\left(-\frac{U(x)}{k_B T}\right) ρ(x)∝exp(−kBTU(x)),势能越低,概率越高。 - 过程的自由能变化是什么?
过程的自由能变化等于终态自由能减去初态自由能: Δ G = G final − G initial \Delta G = G_{\text{final}} - G_{\text{initial}} ΔG=Gfinal−Ginitial,决定了过程的自发方向( Δ G < 0 \Delta G<0 ΔG<0自发)。
Task
课后练习参考:https://github.com/carlocamilloni/Structural-Bioinformatics/blob/main/Notebooks/t02_intro_stat.ipynb
前半部分介绍colab的用法,参考cheatsheet:https://towardsdatascience.com/cheat-sheet-for-google-colab-63853778c093/
一个比较常用的技巧,在cell中执行shell命令
markdown
# 1. 使用magic command
%ls
# 2. 使用 %%bash
%%bash
ls
这种写法下需要注意,bash和python语法不能够混用,
如果注释写第1行,cell会认为这是1个python code,就不能够执行shell脚本了,
所以建议将注释移动到 (%%bash)符合下面
练习部分主要是到pdb数据库下载1个pdb结构文件,然后进行简单的统计分析,
由于比较简单,几分钟过一下code
markdown
#Download a structure from the PDB:
import urllib.request
urllib.request.urlretrieve('http://files.rcsb.org/download/7tjh.pdb', '7tjh.txt')
# here we change the extension of the file from .pdb to .txt so that the computer immediately understand that is a text file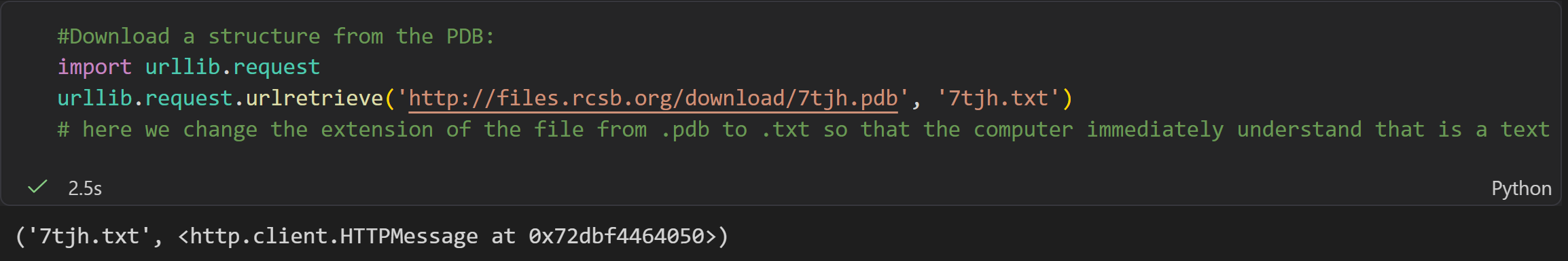
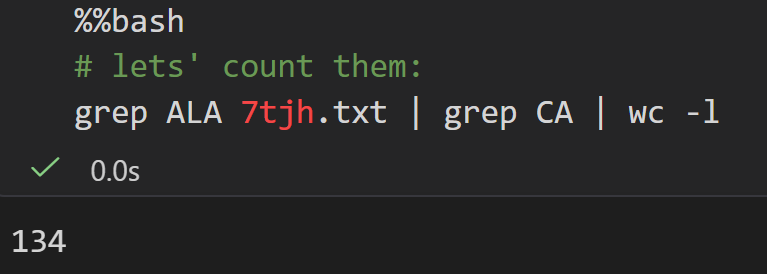
测试Ala丙氨酸
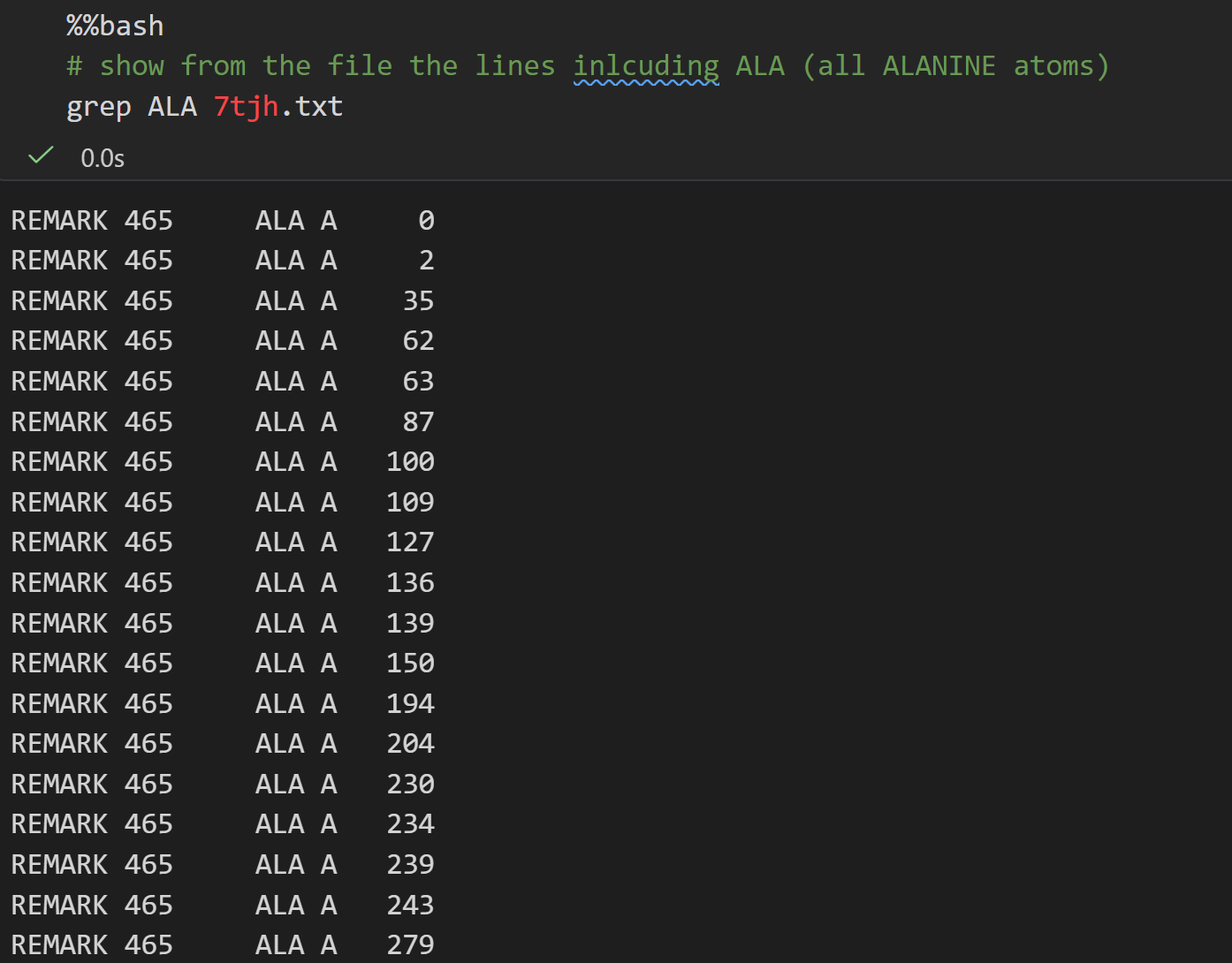
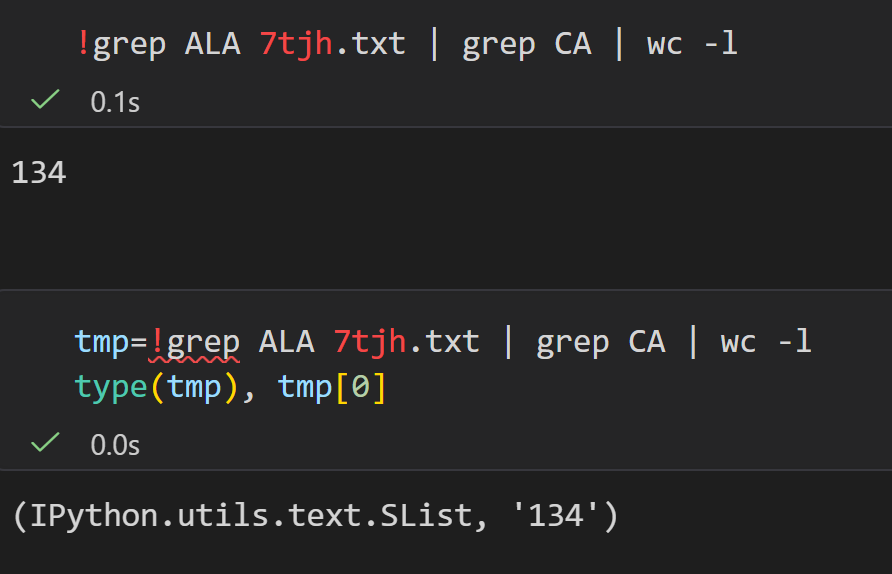
其中具有CA也就是α-C的原子,
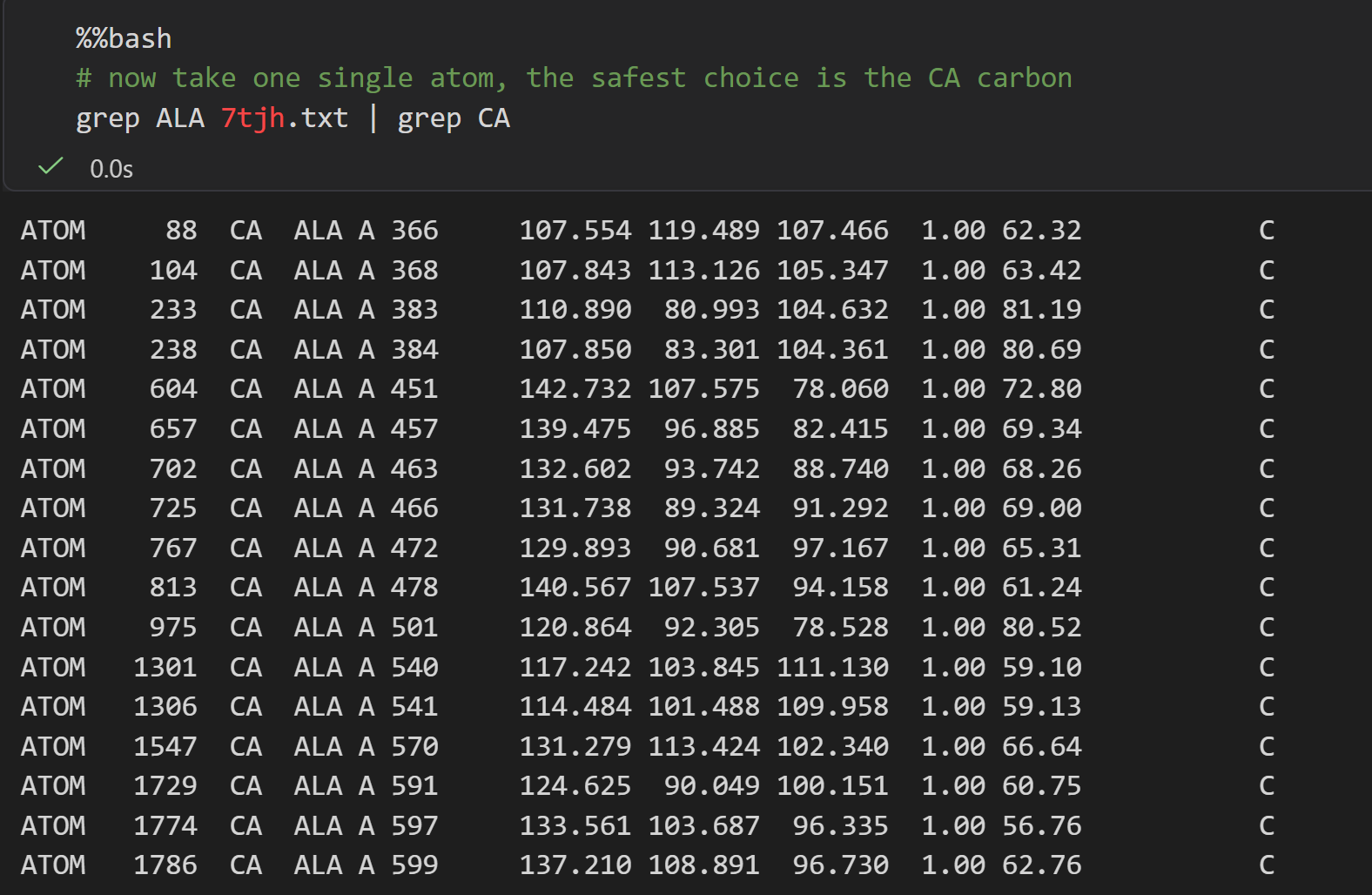
总之我们可以看到,这个7tjh结构中大概有134个Ala的α-C原子,也就是大概有134个Ala
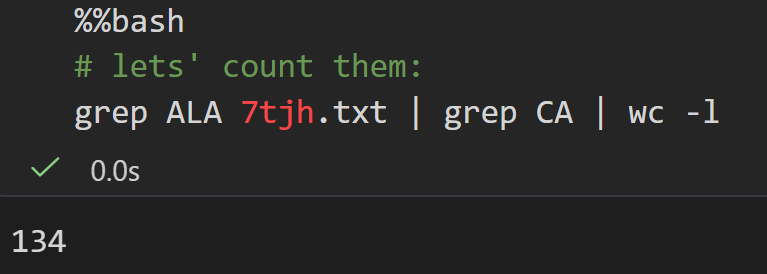
然后注意cell全局shell用(%%bash),局部混用用magic command即可;
另外需要注意的是 !命令返回的是1个列表list,
类型是IPython.utils.text.SList

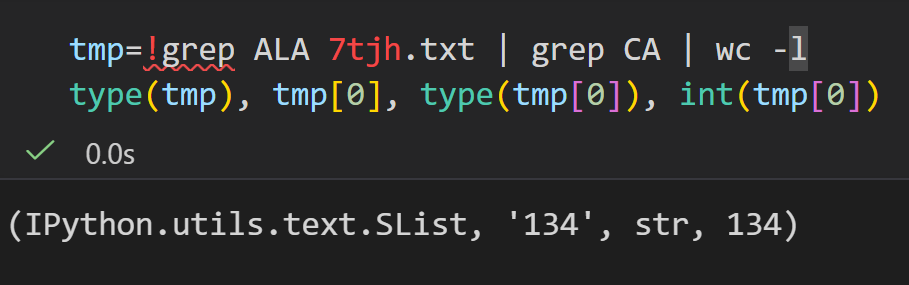
而且注意是字符串列表,文本格式,不是数字
现在我们确定了Ala的数目
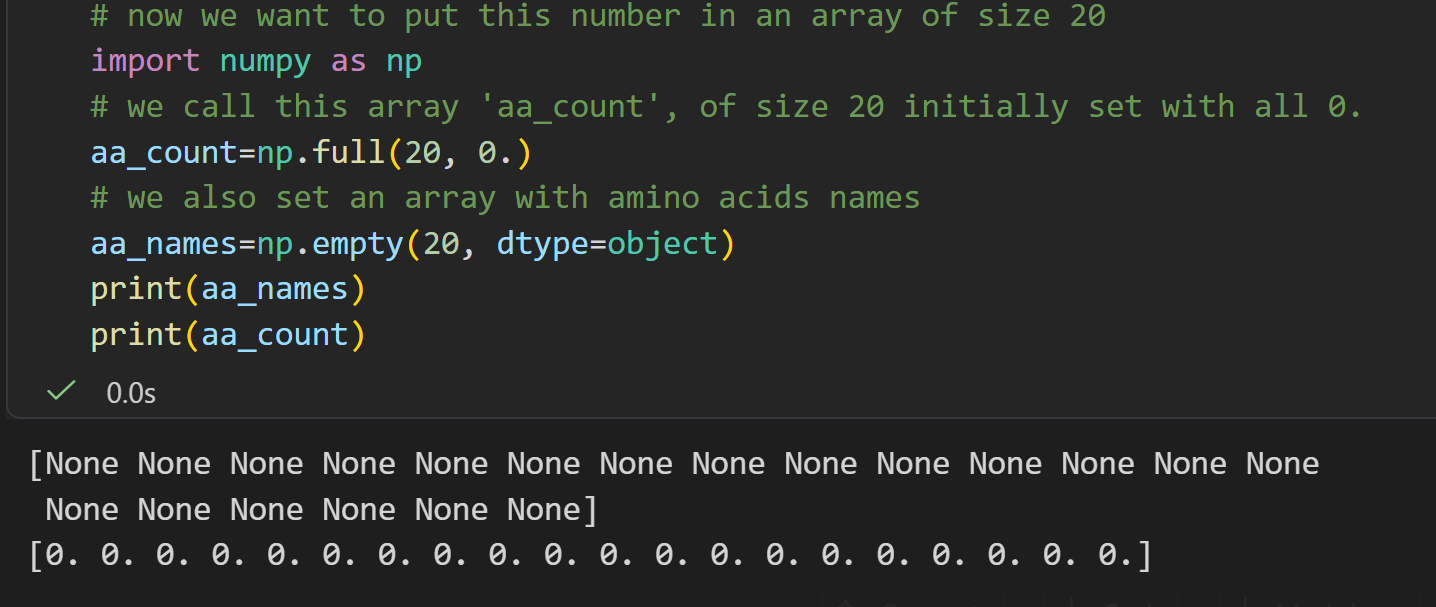
markdown
tmp=!grep ALA 7tjh.txt | grep CA | wc -l
aa_count[0]=tmp[0]
aa_names[0]="ALA"
print(aa_names)
print(aa_count)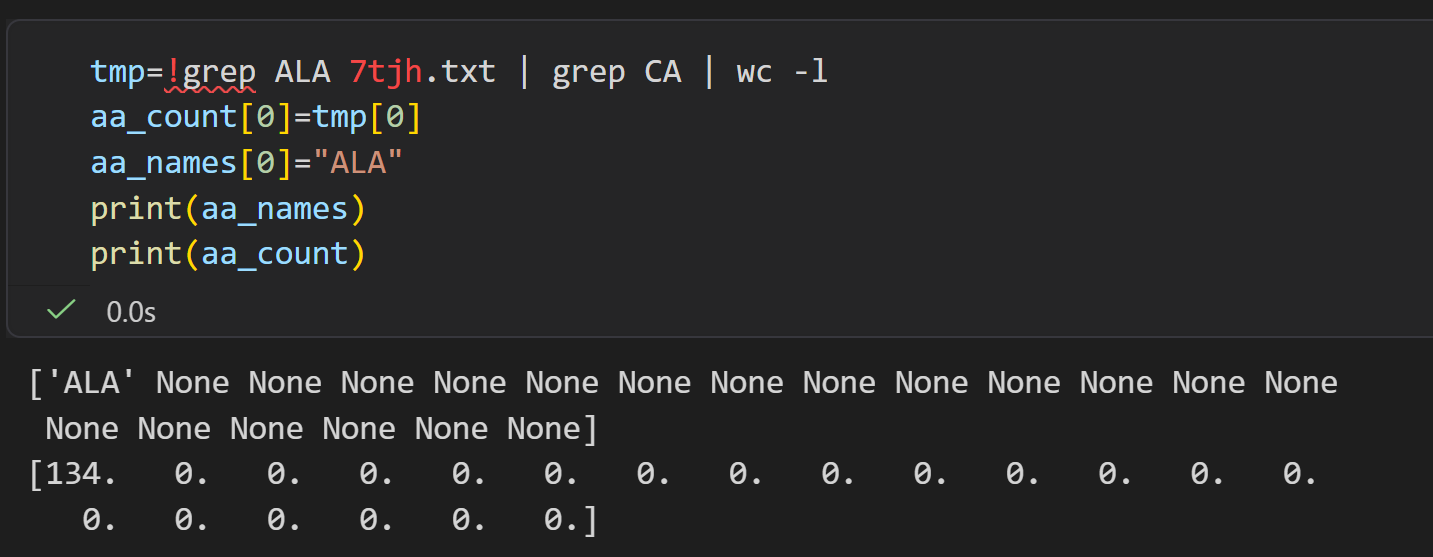
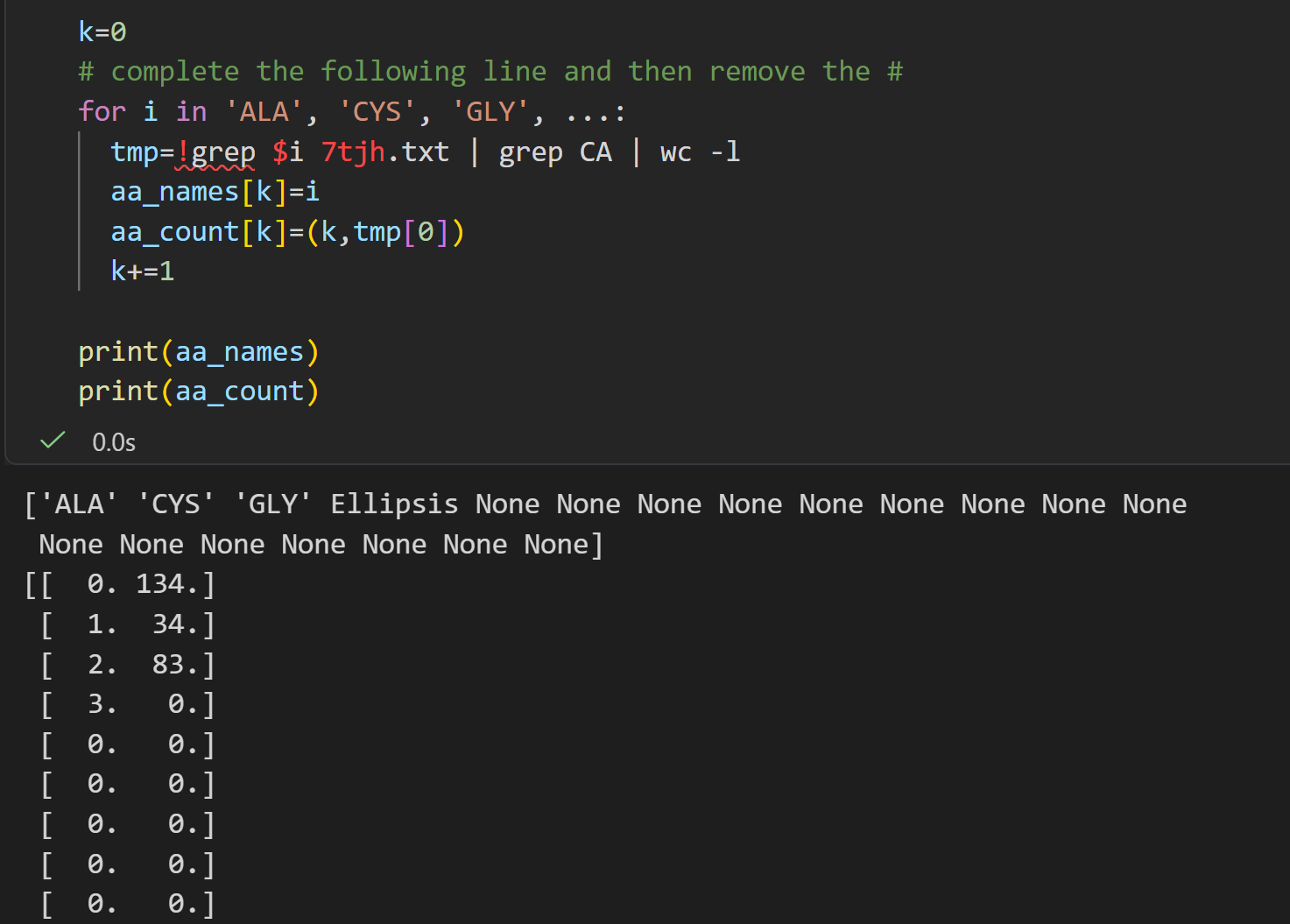
来个循环,
markdown
#we can iterate this operation on some amino acids:
k=0
for i in 'ALA', 'CYS', 'GLY':
print(i)
!grep $i 7tjh.txt | grep CA | wc -l
tmp=!grep $i 7tjh.txt | grep CA | wc -l
aa_names[k]=i
aa_count[k]=tmp[0]
k+=1
print(aa_names)
print(aa_count)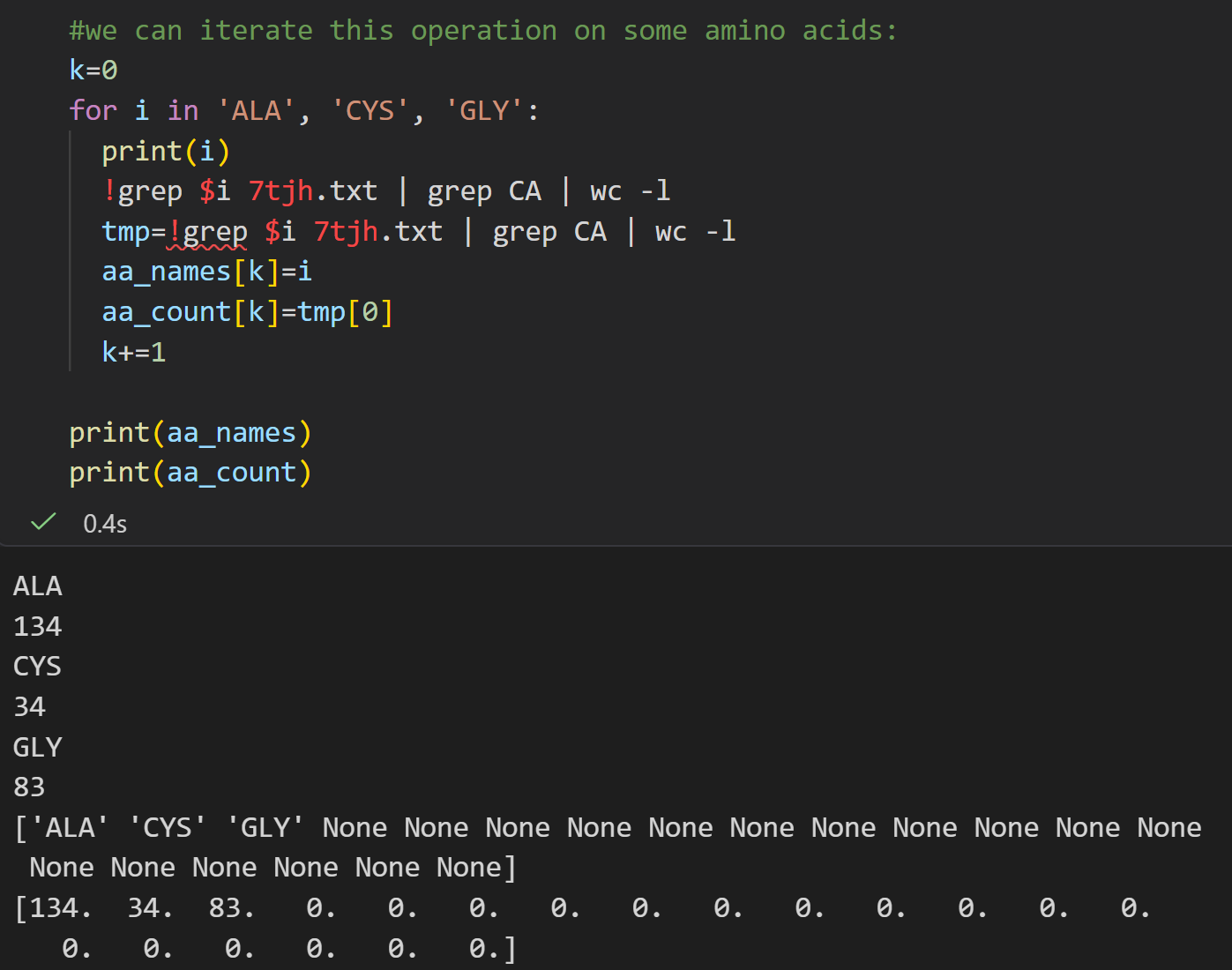
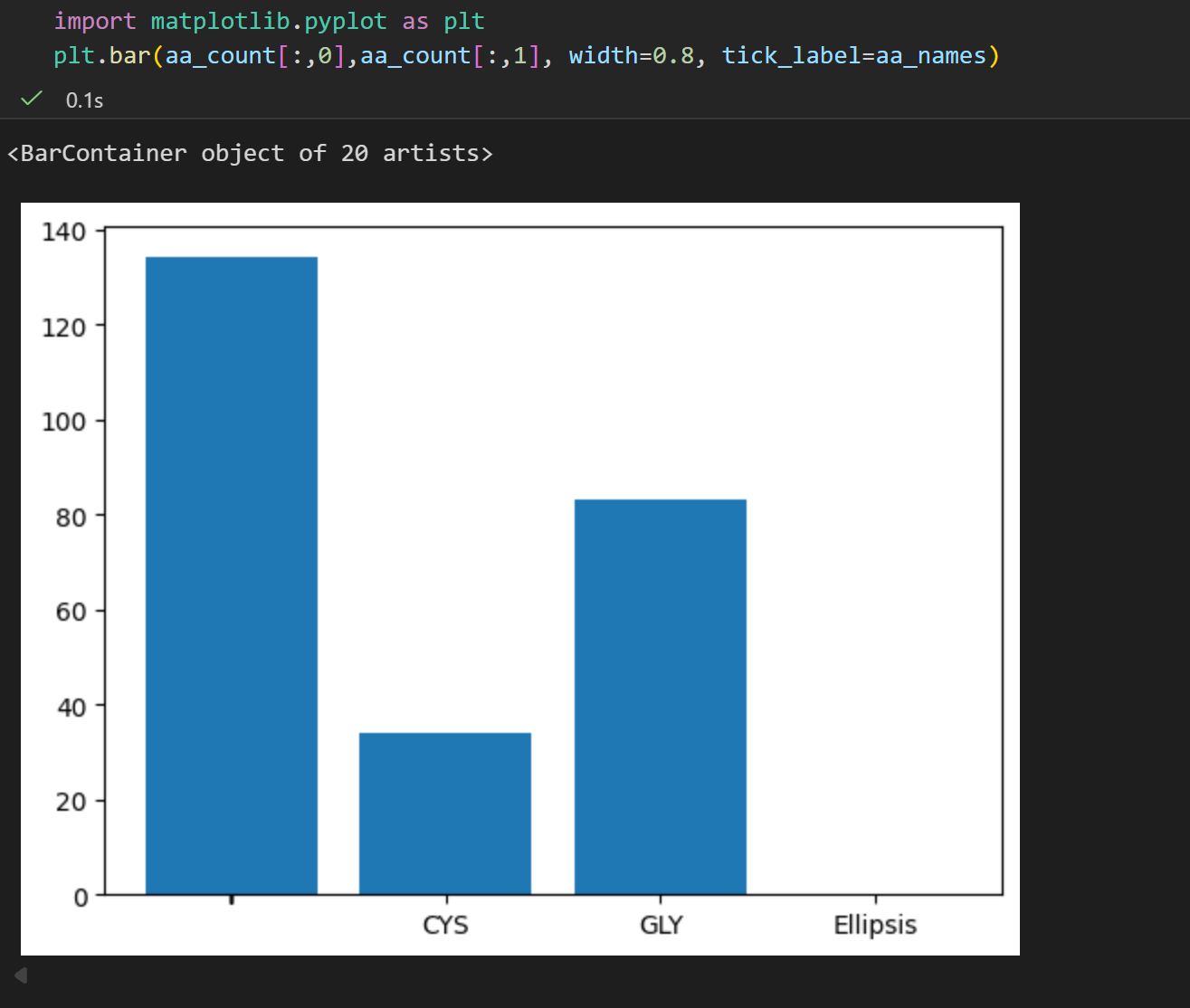
同理,对核酸进行统计,比如说 DNA
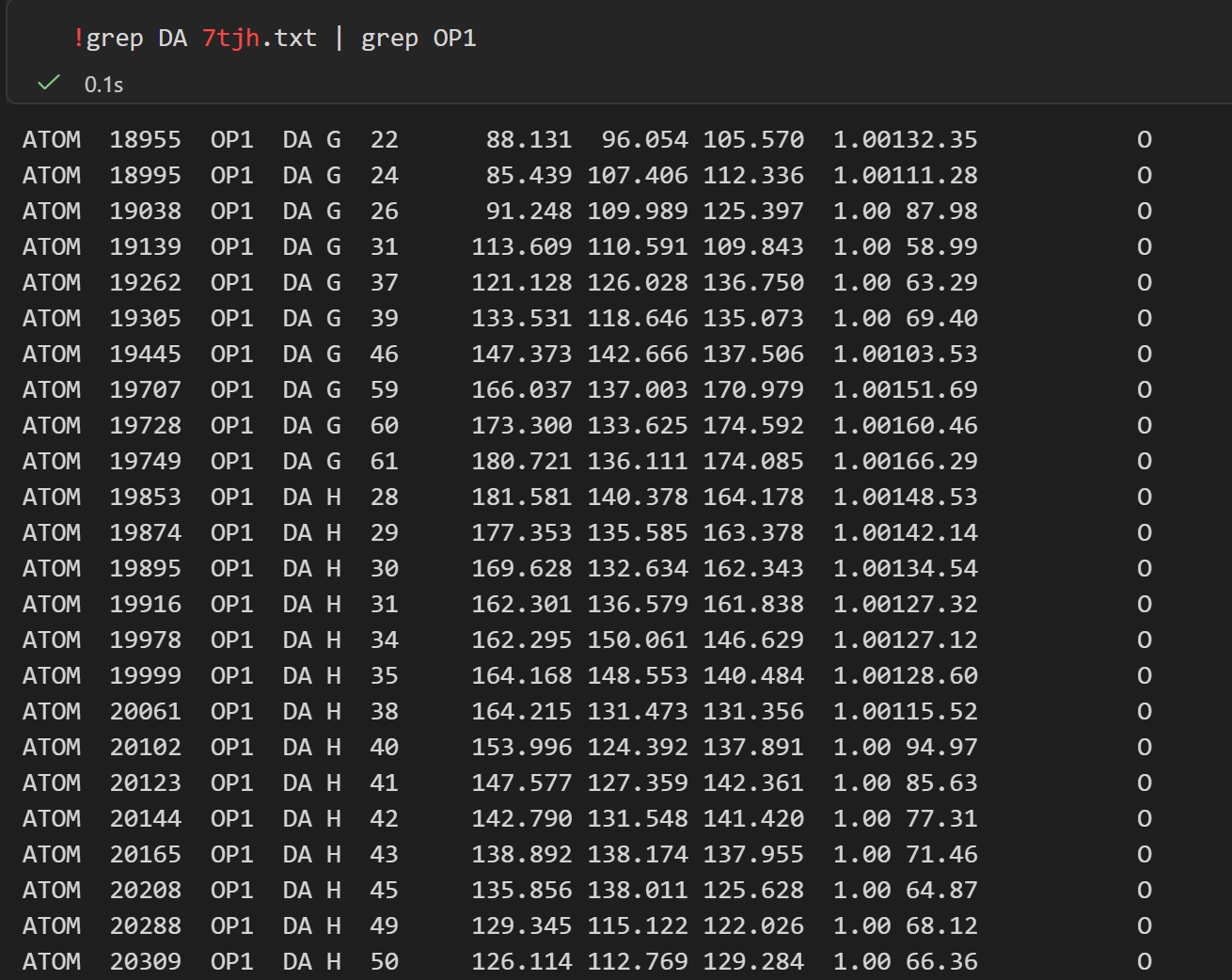
然后是一些简单的统计分析,
用最新的numpy随机数生成函数,

python
# 随机数生成器
rng = np.random.default_rng()
# 带固定随机数种子
rng = np.random.default_rng(seed=42)
# 然后rng.函数方法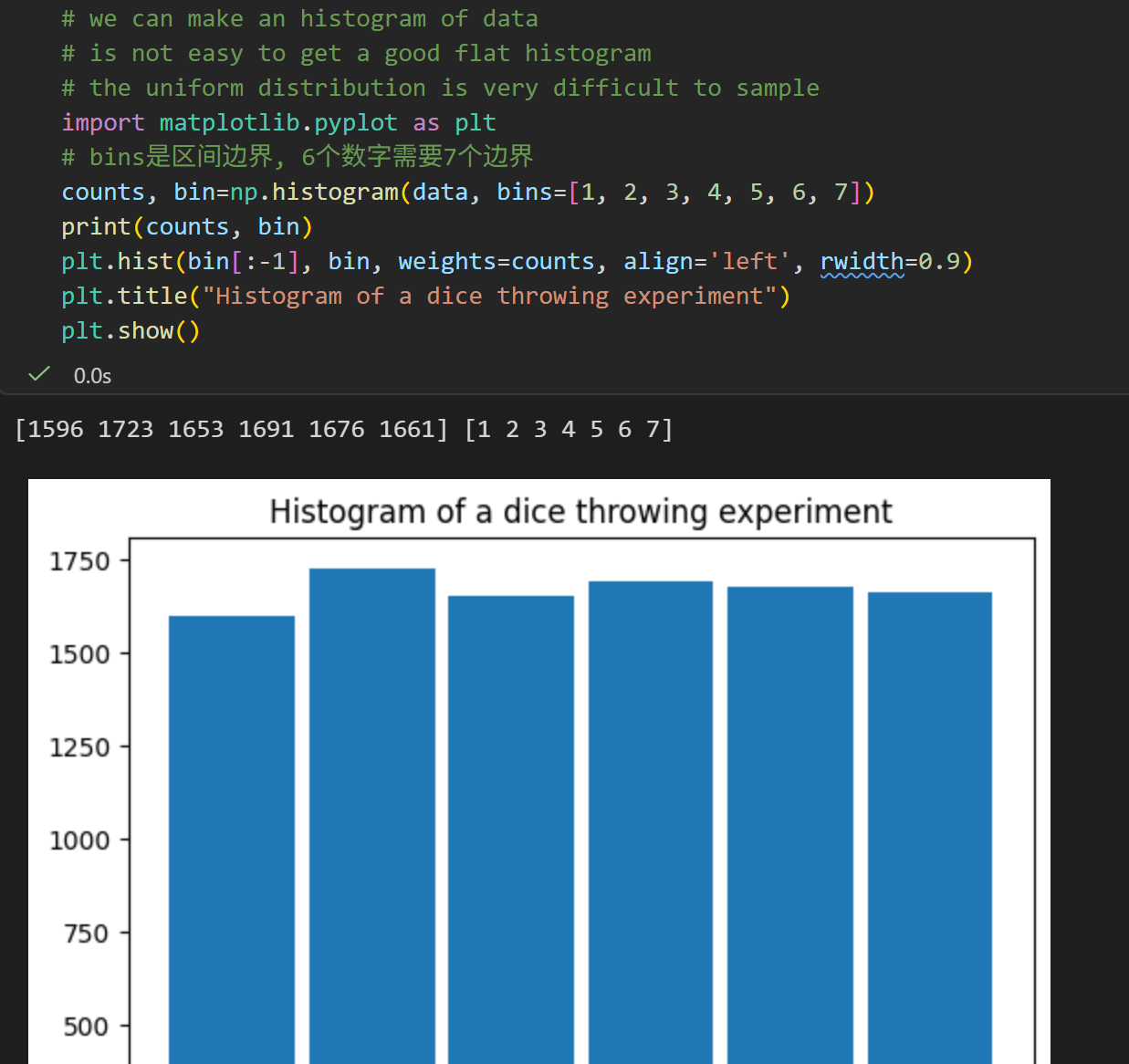
频数归一化成离散的概率密度(两种写法等价)
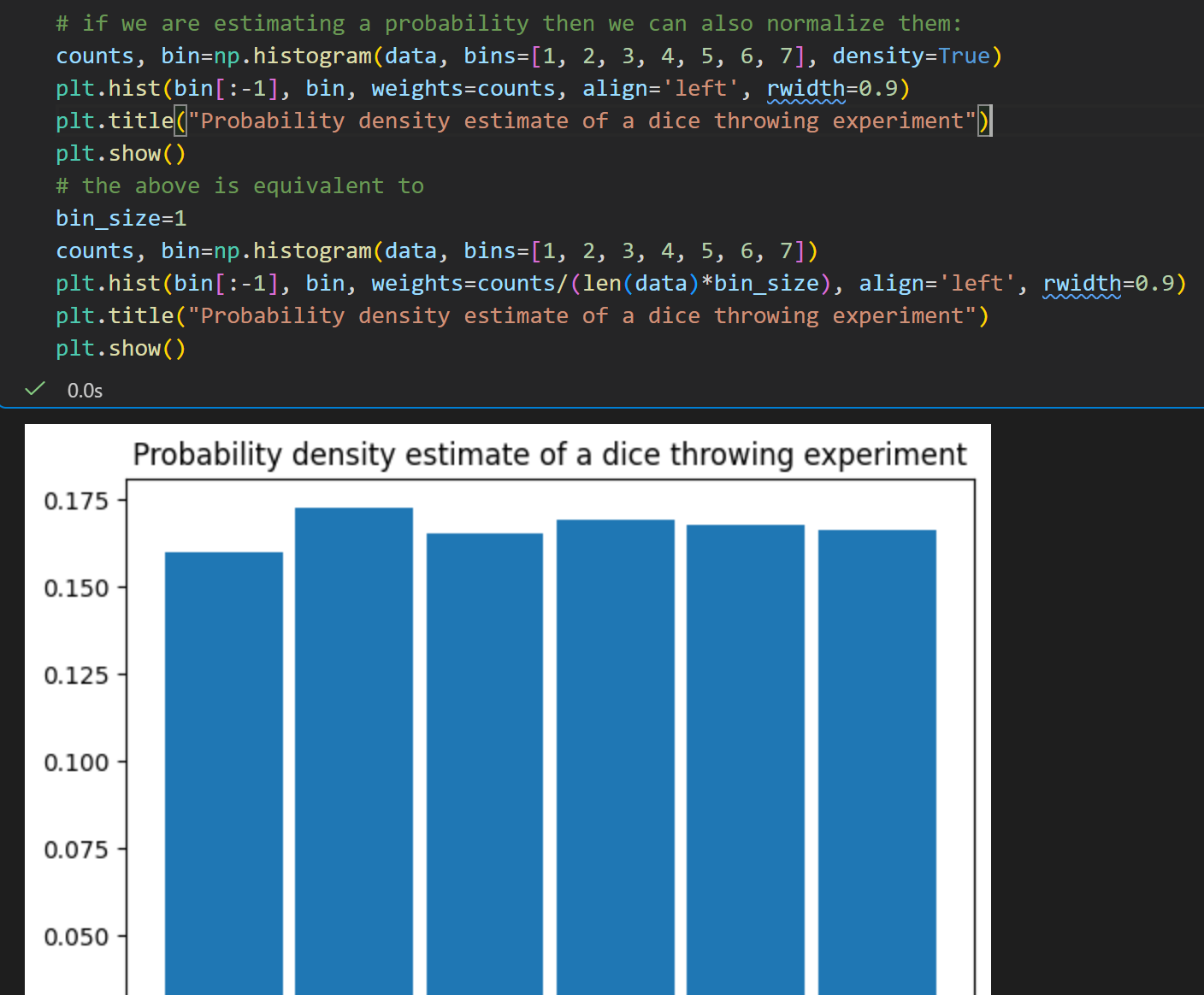
均值与标准差
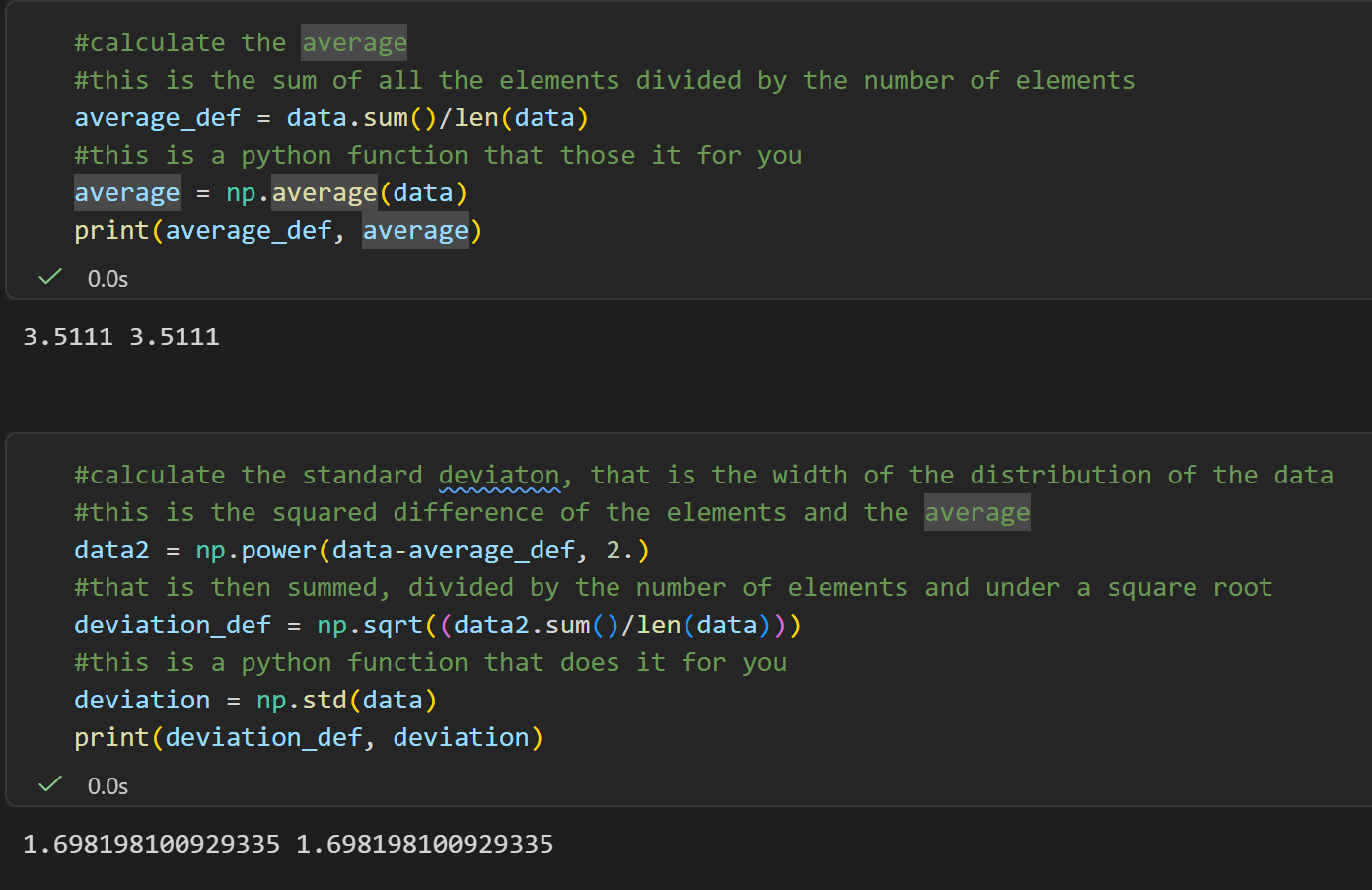
然后是样本标准误,是标准差/(样本数-1)
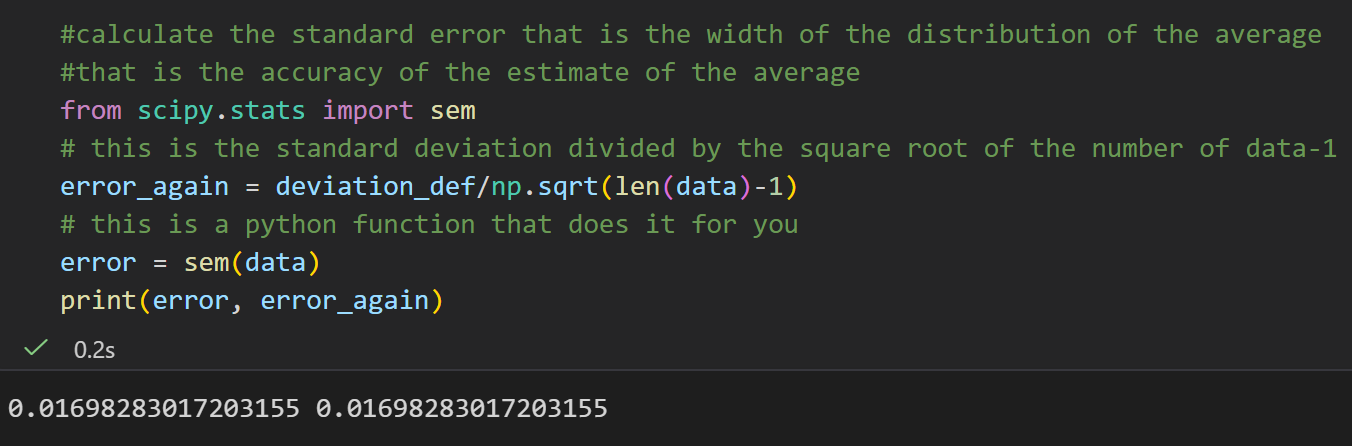

标准误 = 平均值分布的宽度 , 代表平均值估计的精度(越小越准)

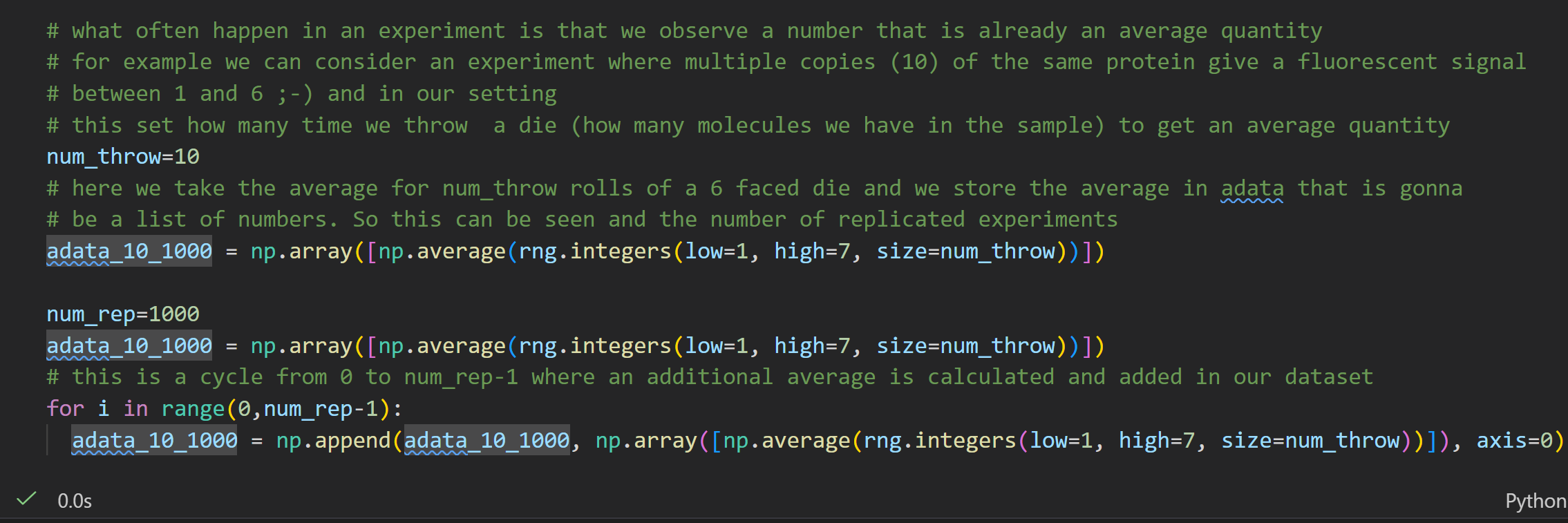
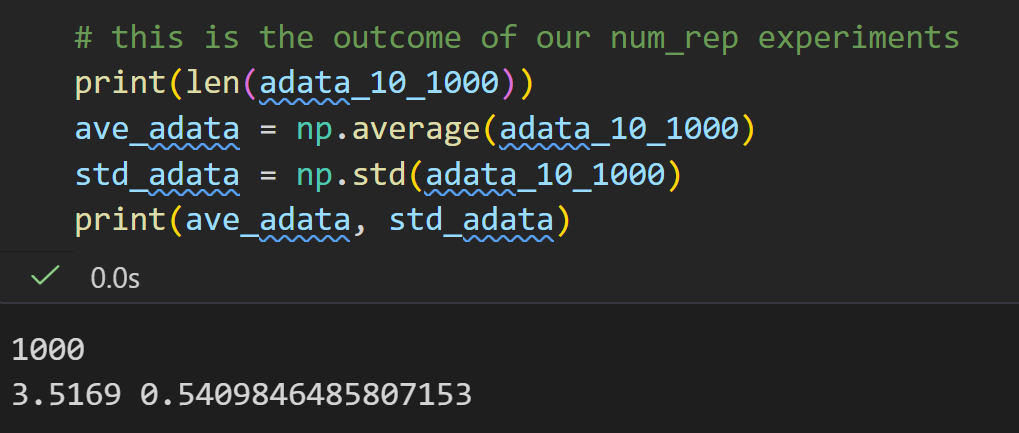
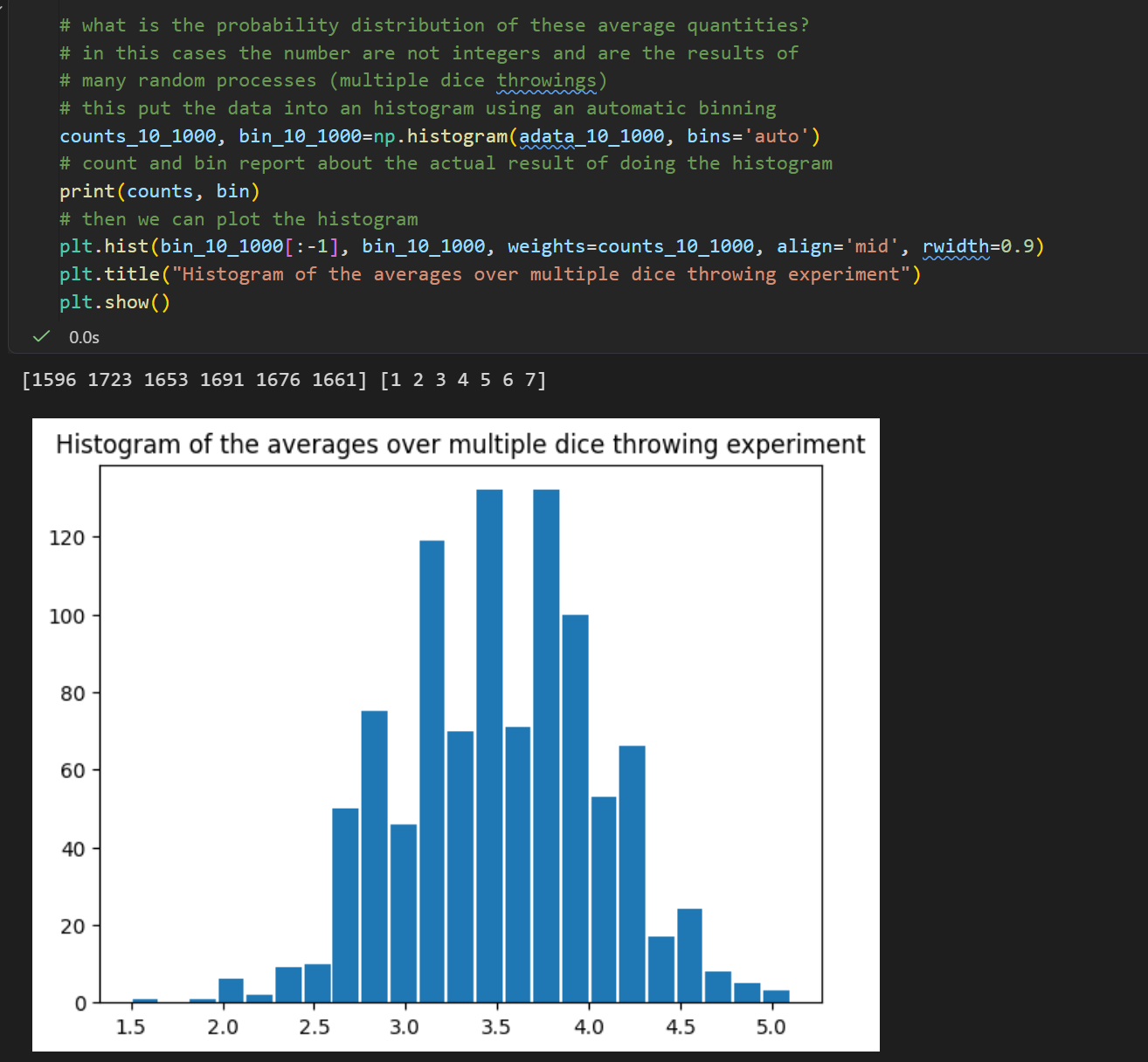
用骰子模拟蛋白荧光读数:单个蛋白荧光 1~6,num_throw=10 = 10 个蛋白为 1 组样本,每组取均值;重复num_rep=1000组独立实验,收集 1000 个组均值存入数组,直观演示实验观测值常本身就是平均值。
生物实验里仪器测出的荧光数值,不是单个蛋白信号,而是多个蛋白信号的平均;代码用骰子等价蛋白信号,批量求平均复刻真实实验读数特征;后续这 1000 个均值的分布会趋近正态分布(中心极限定理)。
中心极限定理就不过多解释了, 不管原始数据是什么分布(均匀、奇怪、乱七八糟), 只要你多次取「平均值」,这些平均值最终一定会变成正态分布(钟形曲线)。


这里大概是在讲我们湿实验测量指标的问题,宏观上其实测量大量分子的平均信号,
然后大量测量的话,得到的这个信号这个表征,其分布必然是正态分布

为什么实验里测到的 "平均值",天然就是正态分布!
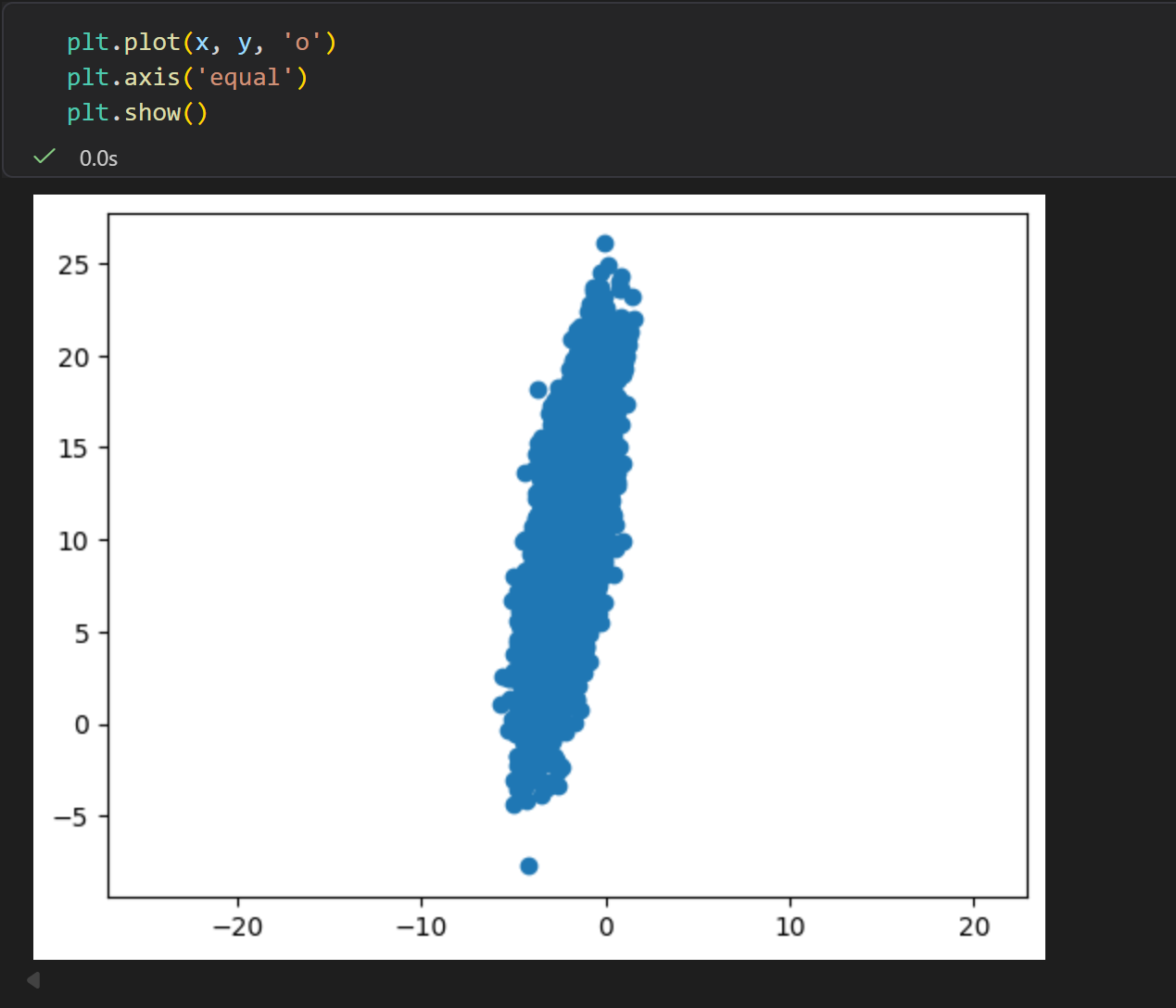
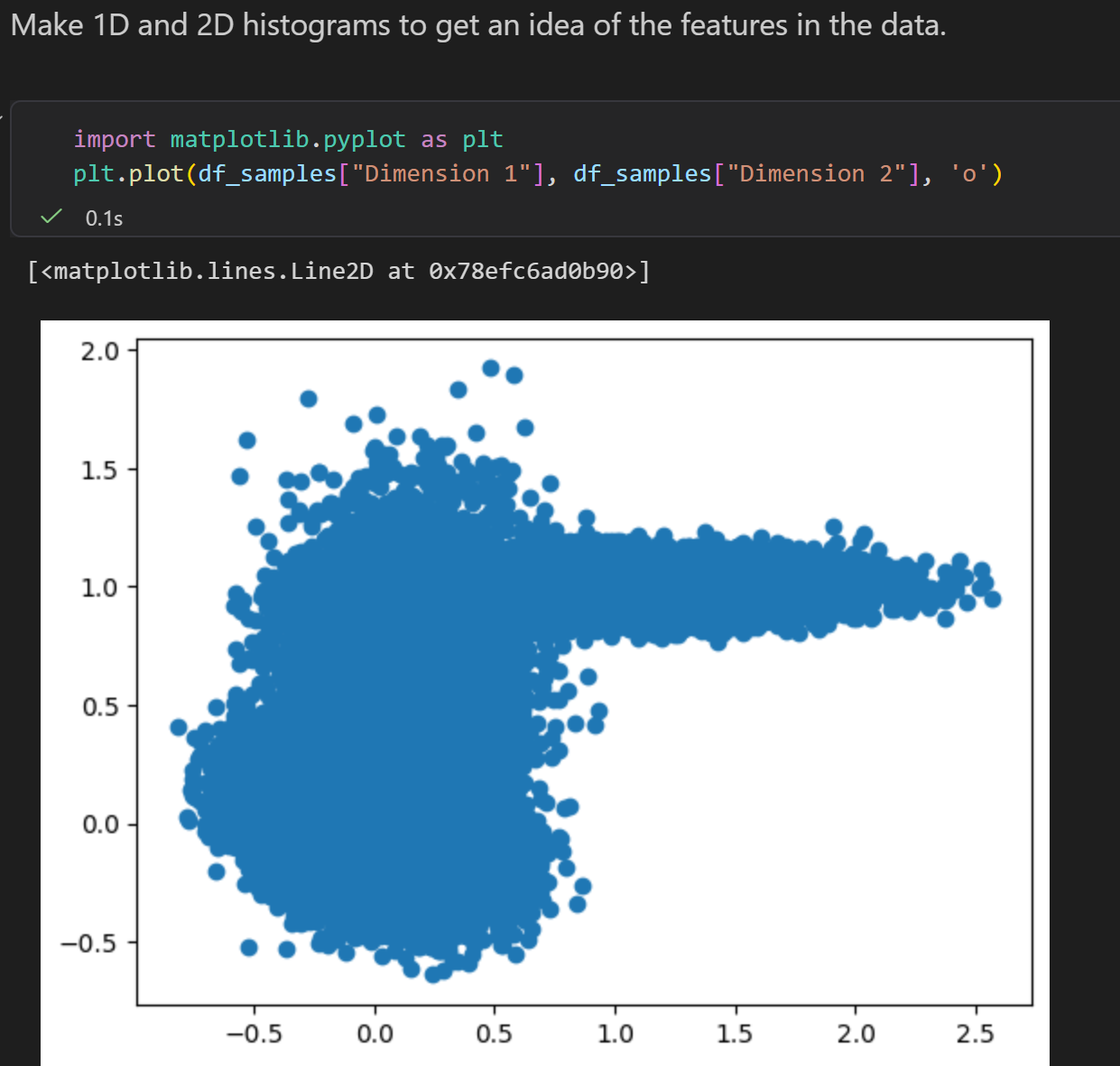
然后是二维分布
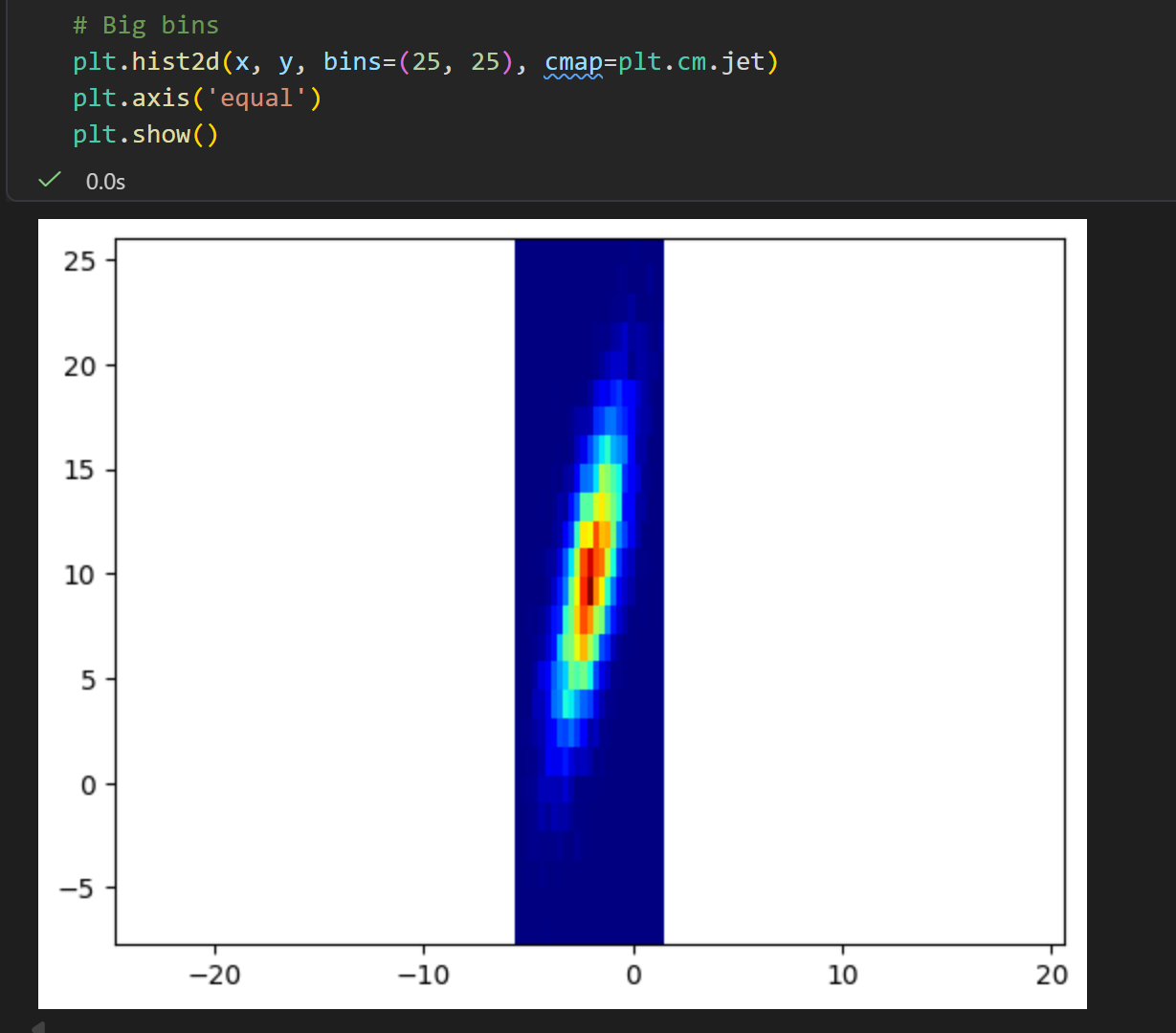
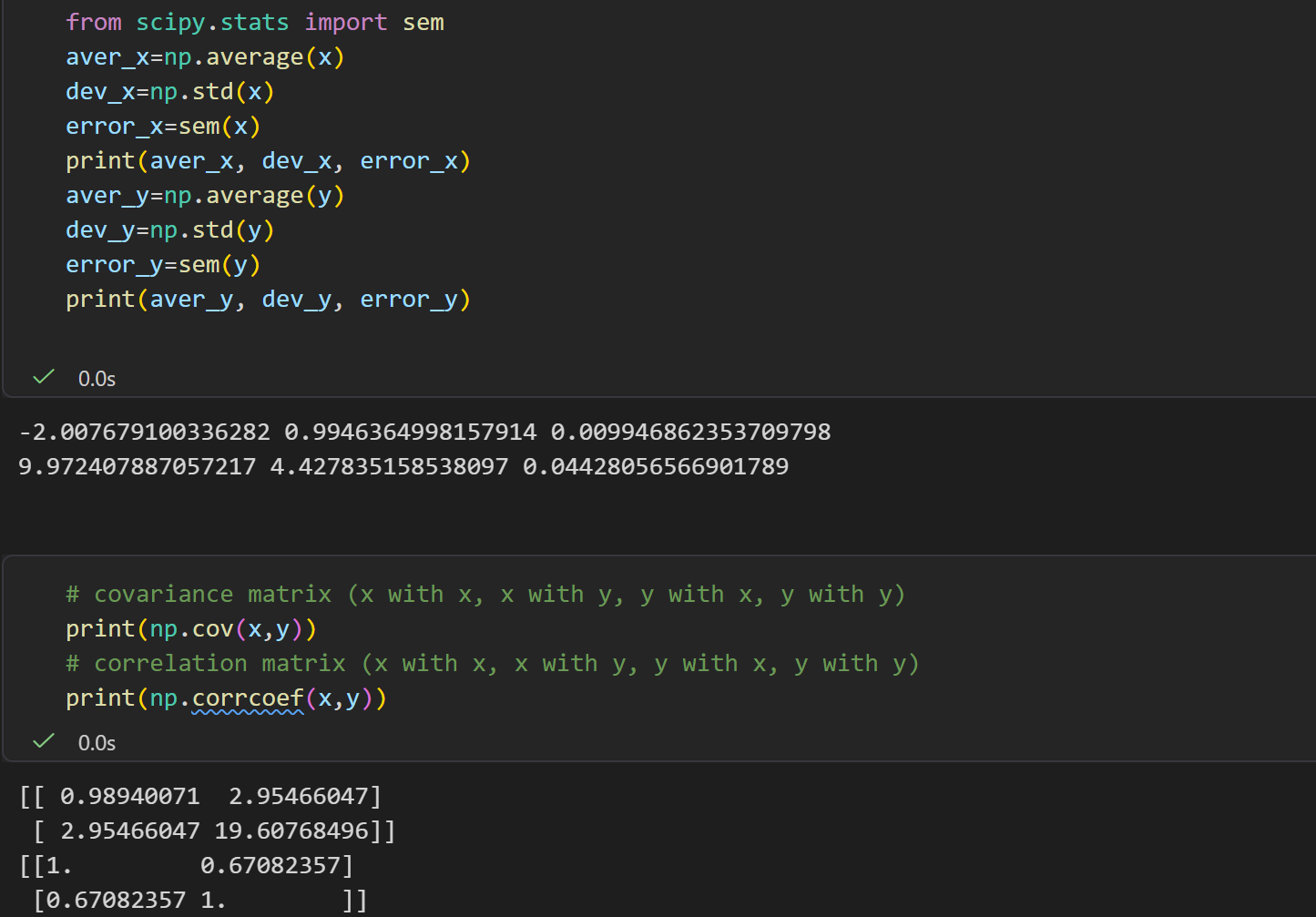
Report
对二维数据
plt.hist2d函数
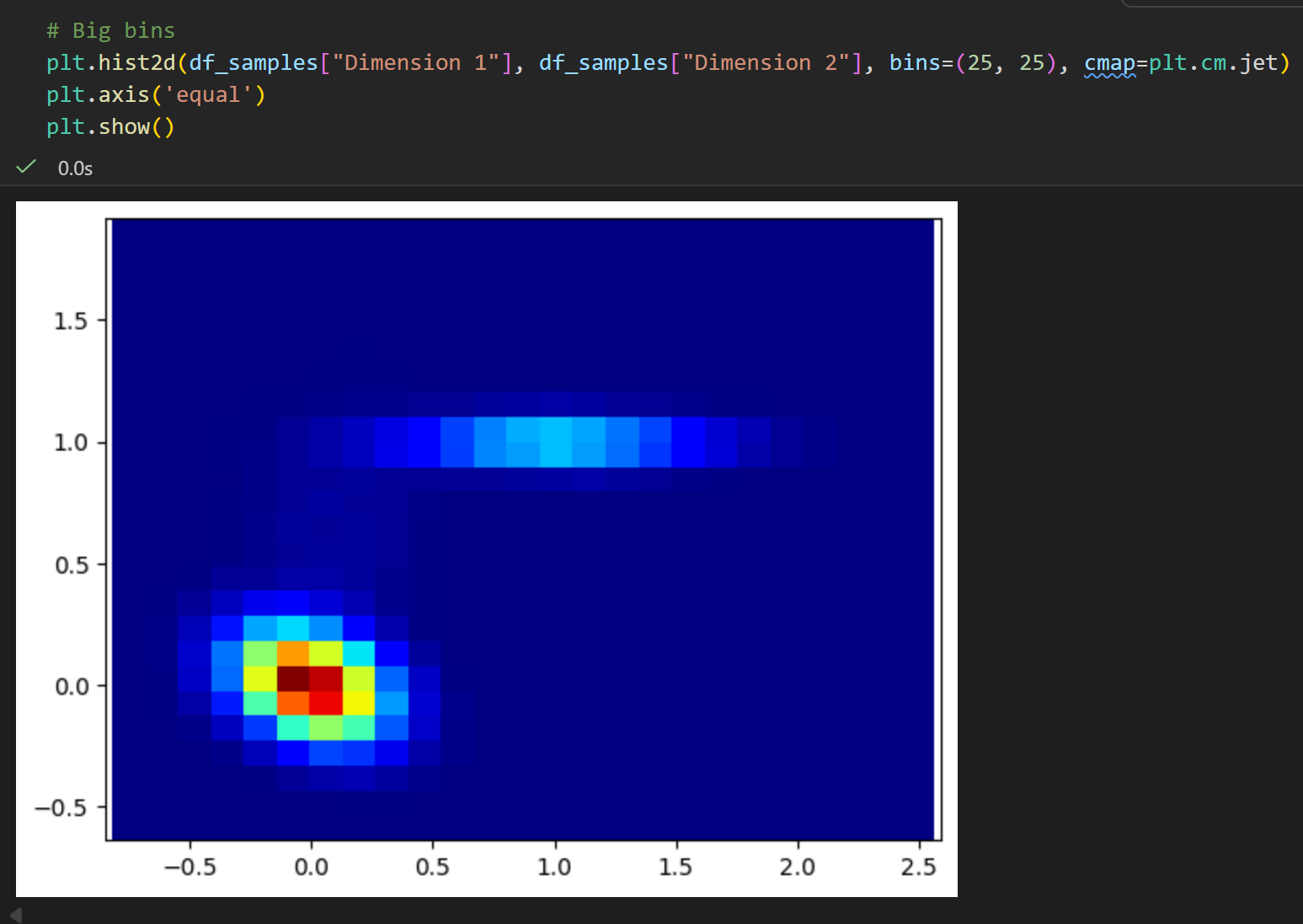
后面这两个提问是不是很摸不着头脑?
我们只有1个二维的分布数据,也就是1个二维的坐标点的长长的lsit/数组,我们只能简单地了解其统计学意义,
做一个简单的二维统计分布,就像前面的分析那样?
但是我们甚至不清楚这些数据的物理意义、物理背景,我们如何联系这里的energy意义?
是不是感觉牛头不对马嘴,完全木有意义?
关键点还是在于前面 统计物理与 概率统计的联系,
也就是概率密度分布已经隐含了能量景观的信息,以及玻尔兹曼分布的公式(能量与概率的关系,我们这里的突破口就是概率,那么我们就能够从概率视角转移到能量视角)。
python
问题是什么,如何理解?
把二维分布直方图 → 转换成 常温(300K)自由能(Free Energy)图,并手动定义两个状态,计算:
✅ 状态的相对布居数(Population)
✅ 状态间的自由能差(ΔΔG)
✅ 绘制专业的自由能等高线 / 热力图⚠️ 下面的分析不一定对,因为没有标准答案,我只提供我个人的一点思考:
- 自由能的本质是「偏好性」
不用测能量,只要看体系喜欢待在哪里(点数多少),就能反推能量。 - 玻尔兹曼分布是通用法则
不管是骰子、蛋白质、原子、实验数据,只要有统计分布,就能算自由能。 - 我们的二维数组 = 分布的数字化表达
它不是普通数字,它记录了数据在平面上的分布规律,这就是自由能的全部信息。
根据玻尔兹曼分布 ,概率分布 ↔ 自由能直接换算:
G = − k B T ⋅ ln ( P ) \boldsymbol{G = -k_BT \cdot \ln(P)} G=−kBT⋅ln(P)
- P P P = 该区域的样本布居数(概率)
- k B T k_BT kBT ≈ 2.479 kJ/mol(常温 300K)
- 自由能越低,代表该区域越稳定
自由能图 + 状态定义 + 自由能差计算:
python
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
# --------------------------
# 1. 基础参数(常温 300K)
# --------------------------
kBT = 2.479 # 单位:kJ/mol,玻尔兹曼常数×温度
bins = (25, 25) # 与前面代码一致的bins
# --------------------------
# 2. 计算二维直方图(计数)
# --------------------------
# 提取二维数据
x = df_samples["Dimension 1"]
y = df_samples["Dimension 2"]
# 计算直方图计数(不绘图,纯数值计算)
counts, x_edges, y_edges = np.histogram2d(x, y, bins=bins)
# 总样本数
total_samples = np.sum(counts)
# --------------------------
# 3. 概率分布 → 自由能计算
# --------------------------
# 转概率(避免0计数导致ln无穷大,加极小值)
prob = counts / total_samples + 1e-12
# 自由能计算(单位:kJ/mol)
free_energy = -kBT * np.log(prob)
# 归一化自由能:最低自由能设为0(最稳定状态为参考)
free_energy -= np.min(free_energy)
# --------------------------
# 4. 绘制二维自由能图
# --------------------------
plt.figure(figsize=(8, 6))
# 绘制自由能热力图
plt.contourf(x_edges[:-1], y_edges[:-1], free_energy.T,
cmap=plt.cm.jet, levels=50)
plt.colorbar(label='Free Energy (kJ/mol)')
plt.xlabel("Dimension 1")
plt.ylabel("Dimension 2")
plt.title("2D Free Energy Landscape (300K)")
plt.axis('equal')
plt.show()
# --------------------------
# 5. 【关键】定义两个状态 + 计算布居数/自由能差
# --------------------------
# 手动定义状态(根据我们的自由能图低谷区域修改阈值!)
# 示例:按 Dimension 1 划分两个状态
STATE1_THRESHOLD = 0 # 状态1:Dim1 < 0
STATE2_THRESHOLD = 0 # 状态2:Dim1 > 0
# 筛选两个状态的样本
state1 = df_samples[df_samples["Dimension 1"] < STATE1_THRESHOLD]
state2 = df_samples[df_samples["Dimension 1"] > STATE2_THRESHOLD]
# 计算布居数(Population)
pop1 = len(state1) / total_samples
pop2 = len(state2) / total_samples
# 计算状态自由能(以最低自由能为0)
G1 = -kBT * np.log(pop1)
G2 = -kBT * np.log(pop2)
delta_G = G2 - G1 # 自由能差:State2 → State1
# --------------------------
# 6. 打印结果
# --------------------------
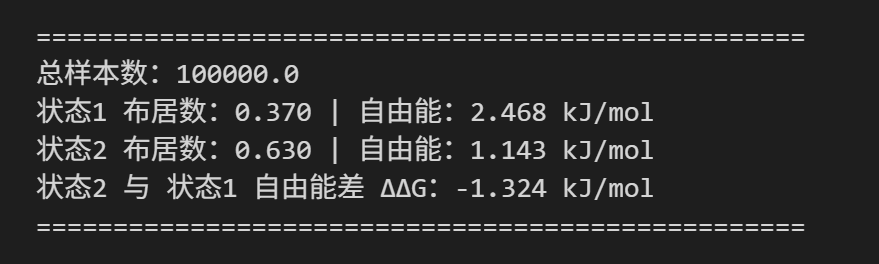
print("="*50)
print(f"总样本数:{total_samples}")
print(f"状态1 布居数:{pop1:.3f} | 自由能:{G1:.3f} kJ/mol")
print(f"状态2 布居数:{pop2:.3f} | 自由能:{G2:.3f} kJ/mol")
print(f"状态2 与 状态1 自由能差 ΔΔG:{delta_G:.3f} kJ/mol")
print("="*50)1. 把直方图 → 自由能图
- 原代码:
plt.hist2d只画计数分布 - 新代码:用
np.histogram2d计算计数 → 转概率 → 算自由能 → 画自由能势能面 - 颜色代表自由能高低:蓝色/深色=低能稳定,红色=高能不稳定
2. 定义状态
此处用 Dimension 1 的数值阈值 划分了两个状态(可以根据自由能图的低谷修改阈值):
- State 1:低能稳定构象
- State 2:高能/另一稳定构象
3. 计算核心物理量
- 相对布居数
该状态的样本数 / 总样本数 - 状态自由能
G = − k B T ln ( P ) G = -kBT\ln(P) G=−kBTln(P) - 自由能差
Δ Δ G = G 2 − G 1 \Delta\Delta G = G_2 - G_1 ΔΔG=G2−G1
自由能不需要复杂的分子模拟/公式,只需要「样本分布」!
我们的二维数组 = 样本的分布计数 → 这就是计算自由能的全部原始所需。
先明确一下我们输入的是什么?
我们这里说的简单二维数组,本质是:
一个二维直方图 :把平面分成很多小格子,每个格子里写一个数字 → 这个数字 = 有多少个数据点落在这个格子里
比如:
- 格子A:100个点
- 格子B:10个点
- 格子C:0个点
这就是统计分布,是计算自由能的唯一输入。
核心物理逻辑就是玻尔兹曼分布,这是底层的灵魂,也是为什么简单分布能算自由能的原因:
统计物理铁律,
数据点出现得越多的地方 → 体系越喜欢待在这里 → 自由能越低(越稳定)
数据点越少的地方 → 体系不喜欢 → 自由能越高(越不稳定)
第1步:把「计数」变成「概率」
- 输入:二维数组(每个格子的点数)
- 操作:每个格子的点数 ÷ 总点数
- 计算意义 :
得到体系出现在这个格子的概率
(概率0~1,概率越高=越喜欢这里)
第2步:概率 → 自由能(核心公式)
- 操作:
自由能 = -kBT × ln(概率) - 计算意义 :
把「概率高低」直接翻译成「自由能高低」- 概率大 → ln(概率) 大 → 自由能 小(稳定)
- 概率小 → ln(概率) 小 → 自由能 大(不稳定)
第3步:自由能归一化
- 操作:所有自由能 - 最小的自由能
- 计算意义 :
把最稳定的地方设为0自由能 (参考点)
其他区域的能量 = 比最稳定状态高多少
第4步:定义状态 → 算能量差/布居数
- 操作:圈两个区域(状态1、状态2),统计里面的总点数
- 计算意义 :
- 布居数:两个状态的占比(谁更常见)
- 自由能差:两个状态谁更稳定,稳定多少
我们这里用最简单的code
python
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
# --------------------------
# 1. 基础参数(常温 300K)
# --------------------------
kBT = 2.479 # 单位:kJ/mol,玻尔兹曼常数×温度
bins = (25, 25) # 与前面原代码一致的bins
# --------------------------
# 2. 计算二维直方图(计数)
# --------------------------
# 提取二维数据
x = df_samples["Dimension 1"]
y = df_samples["Dimension 2"]
# 计算直方图计数(不绘图,纯数值计算)
counts, x_edges, y_edges = np.histogram2d(x, y, bins=bins)
# 总样本数
total_samples = np.sum(counts)
# --------------------------
# 3. 概率分布 → 自由能计算
# --------------------------
# 转概率(避免0计数导致ln无穷大,加极小值)
prob = counts / total_samples + 1e-12
# 自由能计算(单位:kJ/mol)
free_energy = -kBT * np.log(prob)
# 归一化自由能:最低自由能设为0(最稳定状态为参考)
free_energy -= np.min(free_energy)
# --------------------------
# 4. 绘制二维自由能图
# --------------------------
plt.figure(figsize=(8, 6))
# 绘制自由能热力图
plt.contourf(x_edges[:-1], y_edges[:-1], free_energy.T,
cmap=plt.cm.jet, levels=50)
plt.colorbar(label='Free Energy (kJ/mol)')
plt.xlabel("Dimension 1")
plt.ylabel("Dimension 2")
plt.title("2D Free Energy Landscape (300K)")
plt.axis('equal')
plt.show()
# --------------------------
# 5. 【关键】定义两个状态 + 计算布居数/自由能差
# --------------------------
# 手动定义状态(根据我们的自由能图低谷区域修改阈值!)
# 示例:按 Dimension 1 划分两个状态
STATE1_THRESHOLD = 0 # 状态1:Dim1 < 0
STATE2_THRESHOLD = 0 # 状态2:Dim1 > 0
# 筛选两个状态的样本
state1 = df_samples[df_samples["Dimension 1"] < STATE1_THRESHOLD]
state2 = df_samples[df_samples["Dimension 1"] > STATE2_THRESHOLD]
# 计算布居数(Population)
pop1 = len(state1) / total_samples
pop2 = len(state2) / total_samples
# 计算状态自由能(以最低自由能为0)
G1 = -kBT * np.log(pop1)
G2 = -kBT * np.log(pop2)
delta_G = G2 - G1 # 自由能差:State2 → State1
# --------------------------
# 6. 打印结果
# --------------------------
print("="*50)
print(f"总样本数:{total_samples}")
print(f"状态1 布居数:{pop1:.3f} | 自由能:{G1:.3f} kJ/mol")
print(f"状态2 布居数:{pop2:.3f} | 自由能:{G2:.3f} kJ/mol")
print(f"状态2 与 状态1 自由能差 ΔΔG:{delta_G:.3f} kJ/mol")
print("="*50)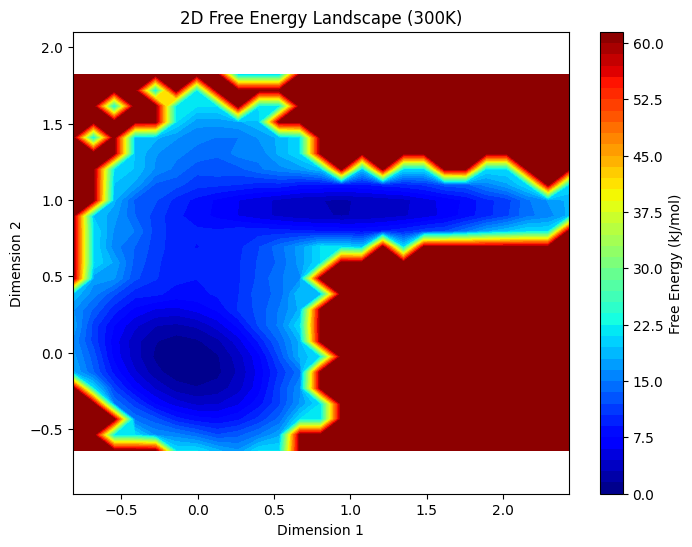
python
import numpy as np
import matplotlib.pyplot as plt
def histogram_to_free_energy(x, y, bins=100, kT=1.0):
"""
将二维散点数据转换为自由能面。
公式: F = -kT * log(P), 其中P是直方图归一化后的概率密度。
Parameters
----------
x, y : array-like, 输入数据
bins : int or tuple, 网格数
kT : float, 温度能量因子
Returns
-------
F : 2D array, 自由能面(相对于全局最小值)
extent : tuple, 绘图范围(xmin,xmax,ymin,ymax)
"""
# 构建2D直方图并归一化为概率密度
hist, xedges, yedges = np.histogram2d(x, y, bins=bins, density=True)
# 处理空网格: 将零概率替换为最小的非零概率值
min_nonzero = hist[hist > 0].min()
hist[hist == 0] = min_nonzero
# Boltzmann反演: F = -kT ln(P)
F = -kT * np.log(hist)
# 将最小值设为0
F = F - F.min()
extent = [xedges[0], xedges[-1], yedges[0], yedges[-1]]
return F, extent
# 应用该函数
F_surface, extent = histogram_to_free_energy(
df_samples['Dimension 1'],
df_samples['Dimension 2'],
bins=100,
kT=1.0
)
# 绘制自由能景观
plt.figure(figsize=(10, 8))
plt.imshow(F_surface.T, origin='lower', extent=extent, aspect='auto', cmap='viridis')
plt.colorbar(label='Free Energy (kT)')
plt.title('Free Energy Landscape from 2D Histogram')
plt.xlabel('Dimension 1')
plt.ylabel('Dimension 2')
plt.contour(F_surface.T, levels=10, extent=extent, colors='white', alpha=0.5)
plt.show()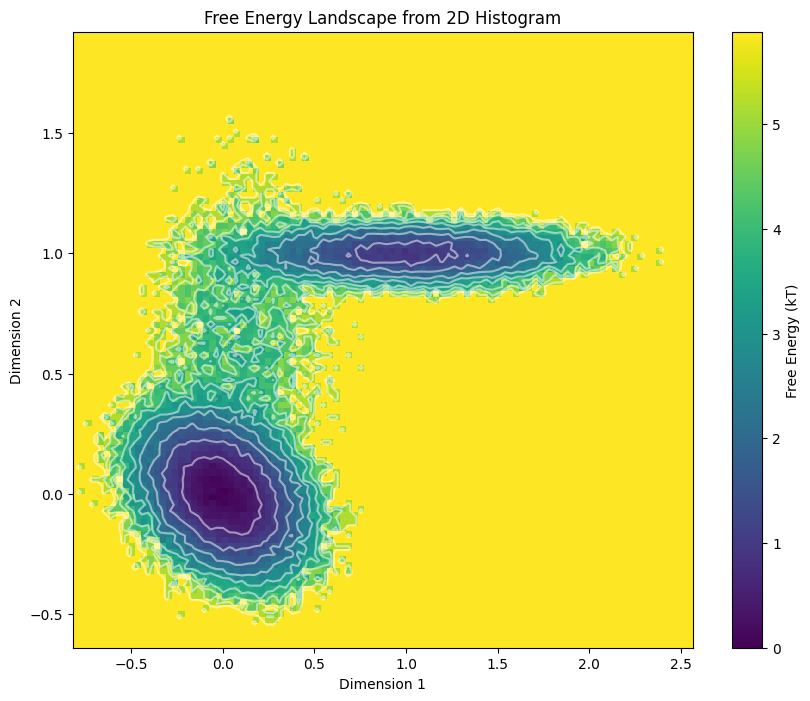
可以用GMM高斯混合model 来选取聚类状态
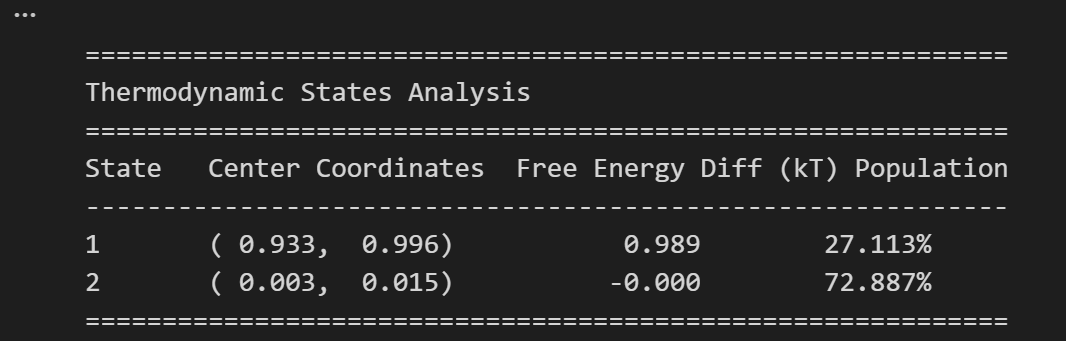
我们再来回顾一下这两张图,
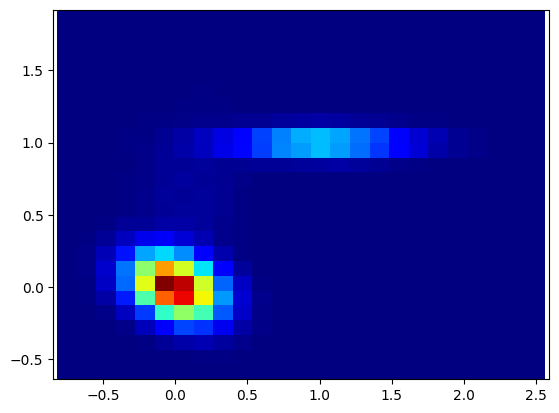
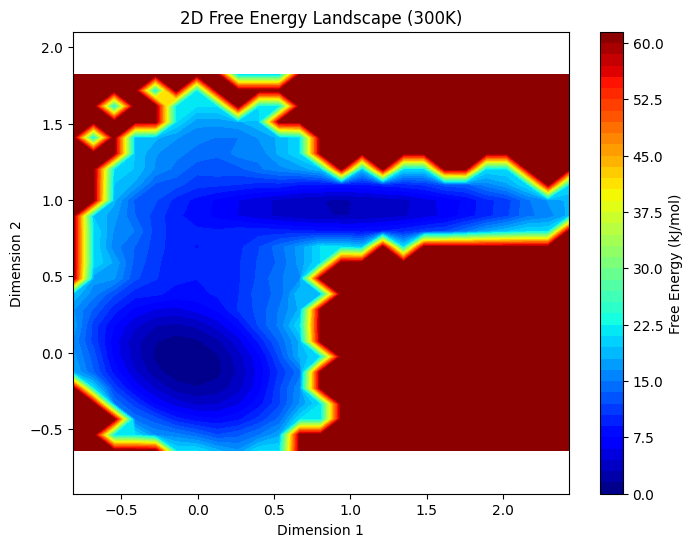
一个是数据分布二维直方图,另外一个是将概率转化为格点中的自由能的二维热图

关键在于有了能量等高线图之后,如何科学定义状态(state)?
我们前面的code只是作为算法演示,给出了最简单的操作:按照0划分。
⚠️ 再次强调,此处我们只是专注于思路、力学与概率的理解,并不确保具体code操作上的正确性。
我们只是演示了随机切分,当然我们需要依托势能面低谷,


补充小问题:能不能随便挑两个单点算 ΔG?
不能。单个格点是单一微观构象 ,而我们研究的 State 是一群相近构象构成的宏观热力学状态;单点能量波动没有统计意义,只有盆地最低能代表整个状态的热力学特征。
所有数据信息都在分布中
- 二维数组 = 数据的分布密度
- 分布密度 ∝ 稳定性 ∝ 自由能高低
- 玻尔兹曼公式 = 把分布直接转成自由能
- 不需要复杂数据!简单分布 = 全部计算依据
最后总结:



这一块应该有类似的程序脚本实现,我们前面是直接使用numpy手搓。



前面提出的两个问题本质上是同一物理量在不同视角下的呈现:概率分布直接反映了占据频率,自由能景观是概率分布经Boltzmann反演后得到的能量地形图。自由能景观可以看作概率分布"取负对数"后的映射,而状态的划分则相当于在这张地形图上识别"山谷"和"盆地"。