NA TURE 丨 使用 AlphaFold 3 准确预测生物分子相互作用的结构
注意!:本文创作仅根据个人理解和网络信息,如有错误恳请指正!谢谢!
大家好,今天分享的文献是2024年5月发表在Nature上的" Accurate structure prediction of biomolecular interactions with AlphaFold 3 "。
1有关作者

Google DeepMind公司是三代AlphaFold 模型的作者,该公司不仅开发了大众熟知的AlphaGo,2016年也将其业务范围从AI+游戏策略拓展到了AI+结构生物学,从2018年在第13届CASP中AlphaFold 1崭露头角到上个月AlphaFold 3可以实现几乎所有生物大分子相互作用的结构预测,可以说DeepMind公司实现了AI在结构生物学领域的重大突破。
2背景介绍
传统地我们利用X射线晶体学、NMR、低温冷冻电镜来进行蛋白质结构的测定,而AlphaFold 2对蛋白质结构的测定就可以达到传统方法所达到的精确度。
AlphaFold 3则不仅仅是对蛋白质结构的预测,更是以一种单一的深度学习框架可以完全预测包含了PDB数据库中几乎所有分子的高精确度结构和相互作用。因此AlphaFold 3为药物研发提供了新的可能性,有望颠覆传统的药物研发模式。
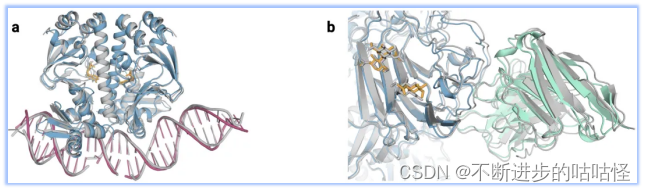
图1|a,DNA和cGMP复合物;b,冠状病毒OC43刺突蛋白;灰色为共晶。
3模型架构
AlphaFold 3的结构是在AlphaFold 2架构基础上进行改进,然后进行训练实现的。AlphaFold 3不仅支持更多种类的分子,同时也提高了训练效率。我们知道,AlphaFold 3可以实现所有所有生物大分子之间相互作用的结构预测,这是因为相较于AlphaFold 2有了更灵活和通用的设计:
-
令牌化方案: AlphaFold3采用了一种更通用的令牌化方案,其中每种类型的分子(如标准氨基酸、核苷酸、其他分子)都对应一个独特的token。对于蛋白质,每个氨基酸残基对应一个token;对于核酸,每个核苷酸对应一个token;对于其他分子,每个重原子对应一个token。这种方案允许模型处理不同类型的生物大分子,而不仅仅是蛋白质。
-
相对位置编码: AlphaFold3引入了相对位置编码,这是一种应用于同一残基内令牌的编码方式,以打破对称性。这种编码方式使得模型能够更好地处理具有对称性的分子结构。
-
输入特征嵌入器: AlphaFold3设计了一个复杂的输入特征嵌入器,它能对所有原子执行注意力操作,从而编码所有分子的化学结构信息,形成一个代表所有token的single representation。这种设计使得模型能够捕捉到不同类型分子的结构和相互作用信息。
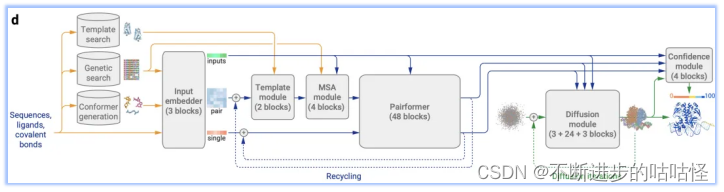
图1d|AF3的架构。矩形表示处理模块,箭头表示数据流向。黄色:输入数据,蓝色:抽象网络激活,绿色:输出数据。彩色代表分子物理原子坐标。
AF3的这种改变在实现了适应各种生物分子的前提下,既简化了模型架构,又保证了性能未受影响。以下为AF3架构的整体流程:
-
模板搜索、基因搜索、构象生成:Genetic search在数据库中搜索与目标序列相似的蛋白质或RNA链;Template search为仅为单链蛋白质链搜索提供模板信息,以辅助结构预测;Conformer generation会根据给定的CCD代码(一种用于标识化学组分的简短字符串编码系统)或SMILES字符(一种用来描述分子结构的字符串表示方法)来生成参考构象。
-
Input embedder:编码所有分子化学结构信息,对所有原子进行Attention,从而输出单表示(single representation)来表示所有的token。同时以类似于AF2的方式构建对表示(pair representation)。然后将该pair对表示和single单表示输入到调节网络(conditioning network),该模块会循环Recycling多次。
-
调节网络:调节网络由Template模块和MSA模块组成,他们分别将有关模板信息和MSA信息编码到pair对表示中,注意这里的MSA信息只针对蛋白质序列和RNA序列。然后生成的pair对表示作为输出,也是下一模块Pairformer模块的输入。
-
单表示(single representation)和调节网络生成的pair对表示输入到Pairformer模块中,形成AF3的主循环。循环后Pairformer模块输出single/pair embedding。
-
扩散模块Diffusion module得到single/pair embedding作为输入进行调节扩散过程,然后输出蛋白质的结构。
-
置信度模块confidence module接收pair对表示、single单表示、从扩散模块得来的结构位置向量来计算置信度度量,如预测的局部距离差异测试(pLDDT)、预测的对齐误差(PAE)、预测的距离误差(PDE)以及实验上已解析的预测等。这些置信度度量有助于评估模型预测的可靠性,并为后续的样本排序和结构选择提供依据。
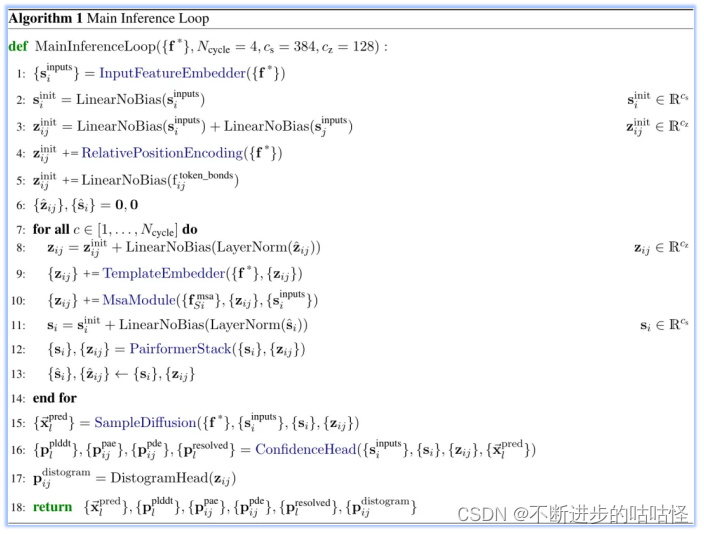
算法1|AF3的整体框架伪代码
3.1输入信息的嵌入(input embedder)
这里是整体流程算法1的第一步(上图),InputFeatureEmbedder。The residue type+参考构象异构体(reference conformer)+MSA摘要特征 (profile and deletion_mean)组合嵌入生成si。The residue type是指输入特征向量的类别标签,它用于指示每个原子属于哪种类型的残基。参考构象异构体reference conformer的生成是由算法5的AtomAttentionEncoder得到。

算法2|input embedder的示意图部分

算法2|InputFeatureEmbedder伪代码
3.2 序列局部原子注意力机制(Sequence-local atom attention)
"序列局部原子注意力"将整个结构表示为原子的平面列表,并允许所有原子在某个序列邻域内直接相互"对话"。例如,每个32个原子的子集关注附近128个原子(在序列编号中邻近)。这使网络能够学习到有关局部结构的语义(补充图1),其中每个标准残基仅用单个token表示。
补充图1|序列局部原子注意力机制。蓝色区域描绘了理论上完整的注意力矩阵。黄色矩形代表实现的局部注意力机制。
3.3MSA模块

补充图2|MSA模块示意图
这里的MSA模块每次循环迭代时会采样MSA的随机子集,然后将采样得到的MSA随机子集和input rep进行嵌入得到msa的单个表示msi。MSA模块有4个同构块,它们进行对表示pair representation和MSA的重复处理和组合。MSA处理后得到的pair representation传递到pairformer模块中。
另外,MSA模块和pairformer模块很相似,其中MSA 表示扮演了类似于单一表示singlerepresentation的角色。相较于AF2这里的注意力对于每一行都是独立执行的,且注意力权重完全从对表示 pair representation中投影而来,无需另外进行键查询注意力。MSA每一行独立的以相同方式进行注意力组合信息也将会减少计算和内存的使用。
MSA注意力层采用与其他注意力层相同的门控机制。否则,模型的这一部分与AF2的工作方式相同,这意味着pair对表示通过三角乘法更新triangle update,和三角自注意力层triangle self-attention,以及过渡块传递。在所有转换块transition中,使用SwiGLU激活函数而不是ReLU。
从概念上讲,这里和AF2的区别在于并不直接组合MSA不同行的信息,所有的信息通过对表示pair对表示流动,动机在于pair对表示应包含尽可能多的有关蛋白质或核酸的信息,因为pair对表示构成了网络其余部分的主干。
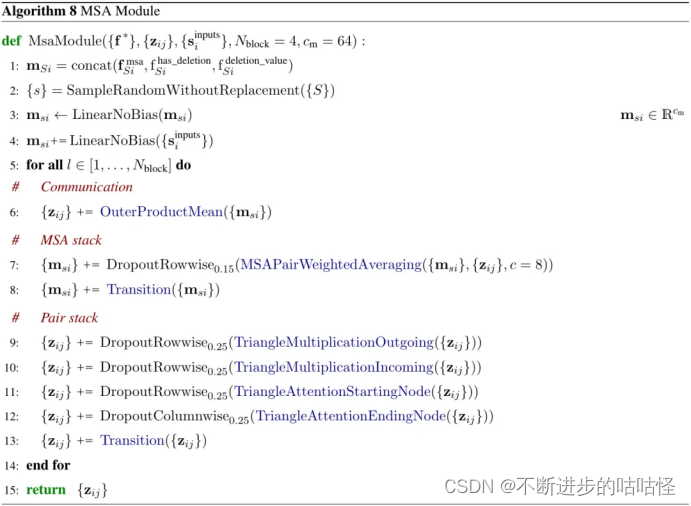
算法8|MSA模块伪代码,zij对表示,msi是MSA表示
3.4 pair对表示的三角形更新
这个pair对表示的三角形更新是MSA模块中的子模块,涉及对成对表示(pair representation)进行特定的更新操作。这些更新旨在捕捉成对节点(例如蛋白质或RNA链中的相邻原子)之间的空间关系,并确保这些关系在整个网络中得到一致且准确的表示。和AF2中的一致,具体细节可以参考AF2论文。
3.5模版模块(Template embedding)
模板嵌入(算法16)将所有原始模板特征组合成一个对表示uij,并将其与前面对表示 zij(在之前的回收迭代中产生)一起处理。这允许网络根据其当前对结构的认识来关注模板中的特定区域。

算法16|模版模块示意图,橙色是搜到的模版,蓝色实线是最开始嵌入embedding得到的pair对表示,蓝色虚线是回收的pair对表示

算法16|模版模块伪代码,最后输出对表示uij
3.6Pairformer模块
Pairformer模块(算法17,图2a)与AF2中的Evoformer是类似的作用,区别在于Pairformer使用单表示si,而不是MSA表示。在这里,single单表示扮演的角色类似于 AF2中Evoformer中的特权第一行。
这个变动,使得不再存在列的注意力(column-wise attention)。具有成对偏差的单一注意力(single attention)与AF2中使用的逐行注意力(row-wise attention)相同,仅作用在单个序列。
此外,与AF2不同的是,single单表示不会影响pair对表示,但pair对表示通过偏置注意力逻辑(single attention with pair bias)来控制single单表示中的信息流。所有转换块transition均使用SwiGLU激活函数。输出的pair对表示和single单表示被传递到后续的扩散模块,该模块取代了AF2的结构模块。
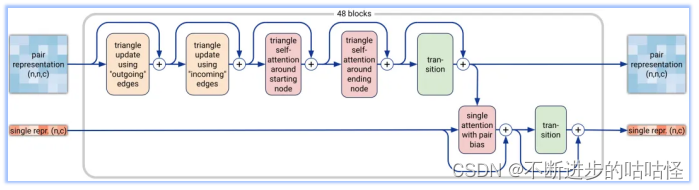
图2a|Pairformer模块示意图,输入输出为pair对表示和single单表示,n为标记数量(聚合物残基和原子),c代表channel数量(对表示为128,单表示为384)。48个blocks中每一个都有一组独立的可训练参数。

算法17|Pairformer模块的伪代码,其中zij是pair对表示,sij为single单表示。
3.7扩散模块
扩散模块直接作用在原始的原子坐标和一个粗略的抽象令牌表示上。这个模块不依赖于旋转框架或任何等变处理。在AlphaFold2中,结构模块需要处理氨基酸特定的帧和侧链扭转角,而扩散模块则通过训练来学习蛋白质结构在不同长度尺度上的信息。在推理阶段,模型会随机采样噪声并反复去噪,以生成最终的结构。这种方法是一种生成式训练过程,它能够产生一系列可能的答案。因此,即使网络在某些位置上不确定,对于每个答案,局部结构也会被清晰地定义出来,例如侧链键的几何形状。这允许模型避免使用基于扭转的残基参数化和违反结构的损失,同时处理一般配体的完整复杂性。
同时作者发现分子的全局旋转和平移不需要不变性或等变性。因此使用扩散模型以简化深度学习架构。扩散模型在训练过程中,是训练一个去噪器(神经网络),以消除以分子骨架的所有重原子位置的高斯噪声。去噪器是基于tranformer,做了些修改如下:
- 多种方式condtioning:应用Adaptive Layernorm的变体到single embedding;应用logit biasing到pair token conditioning。
- 使用标准的现代Transformer(例如:SwiGLU)和 AF2 的门控机制。
- 使用两级架构,首先处理原子,然后处理tokens,然后再次处理原子。
- Transformer仅使用单个线性层来嵌入所有原子位置,并使用单个线性层来投影最后的更新,不涉及几何偏差(例如:局部性或SE3不变性)。

图2b|输入,per-token表示(绿色是inputs表示;蓝色是pair对表示,橙色是single单表示);per-atom condition。输出,彩色球代表物理原子坐标。序列局部注意力机制 (seq.local attention)在前文(补充图1)已介绍。
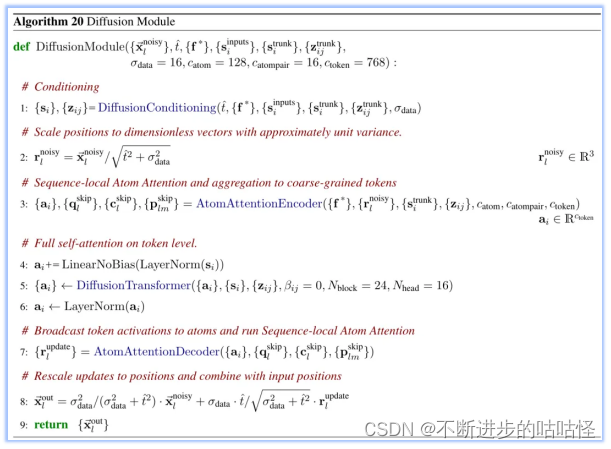
算法20|扩散模块的伪代码
不过扩散模型的使用也会带来问题,最大的问题是生成模型容易产生幻觉,模型将非结构化区域也会输出看似合理的结构(如下图右上角结构)。为了解决此问题,作者使用了一种新颖的交叉蒸馏方法,用AF-Multimer v2.3预测结构来丰富训练数据。在这些结构中,非结构化区域通常是长loop,而非紧凑结构。用这种自蒸馏数据"教导"AF3模仿这种行为。这种交叉蒸馏大大减少了AF3的幻觉行为(扩展图1)。
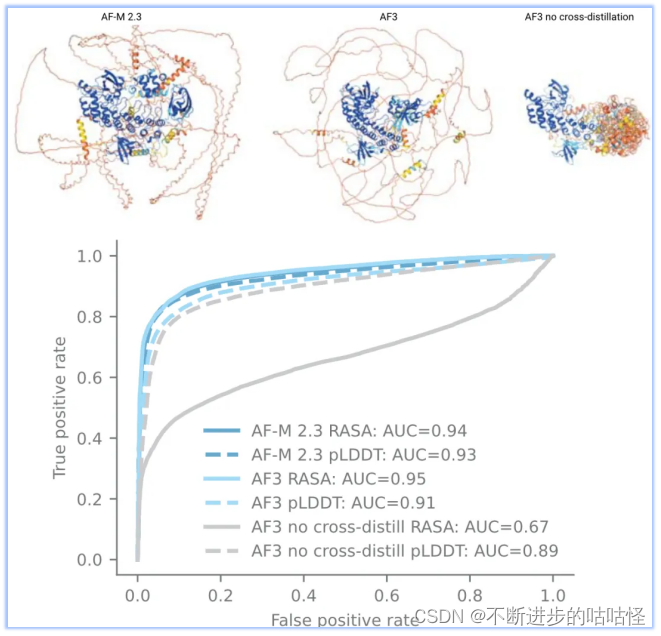
拓展图1|无序区预测。来自AF Multimer v2.3、AF3、没在无序蛋白交叉自蒸馏训练的AF3版本,进行结构预测;蛋白结构按pLDDT着色,蛋白来自CAID2数据集。CAID2 数据集,各种版本AlphaFold的表现。其中RASA是相对可及表面积。
3.8模型置信度模块
结构的置信度,AF2是通过对训练期间结构模块的输出误差进行回归来实现的。然而,该方法不适用于AF3的扩散训练,因为扩散的每一步都会用来训练,而不是直接完整的结构输出(图2c)。于是AF3开发了完整结构预测生成的扩散"推出"机制(使用比正常情况更大的步长)。
然后,该预测结构用于置换对称的真值链和配体(permute ground truth),并计算性能指标(Metrics)以训练置信度模块。置信度模块使用pair对表示来预测pLDDT、对齐误差PAE矩阵、以及距离误差矩阵PDE误差。更多置信度模块的细节原理读者请阅读SI方法4。

图2c|训练设置从网络主干(Network trunk)的末端开始。绿色是inputs表示;蓝色是pair对表示,橙色是single单表示。蓝色箭头,抽象激活数组abstract activation arrays;黄色箭头是真实数据;绿色箭头是预测数据。Stop标志,停止梯度运行。训练和infer两个扩散模块共享权重。
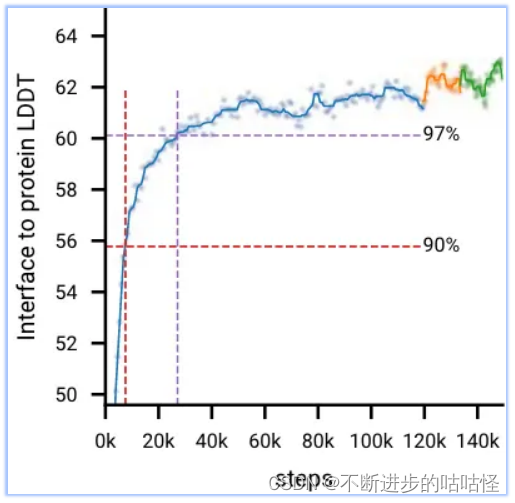
图2d显示,在初始训练期间,模型快速学习预测局部结构(所有生物分子LDDT指标快速上升,并在前20k训练步骤内达到最大性能的97%),而模型需要相当长的时间来学习达到全局收敛(LDDT指标缓慢上升)。在AF3开发过程中,作者观察到一些模型也会较早达到顶峰并开始下降;很可能是由于过度拟合有限数量的训练样本,比如下图protein-rna曲线。

图2d|初始训练和微调阶段的训练曲线,十字标记了达到训练最大值97%的点。
怎么解决上面指出的过拟合问题?
- 通过增加/减少相应训练集的采样概率,
- 以及使用上述所有指标的加权平均值和一些附加指标提前停止,来选择最佳模型checkpoint。
- 在微调阶段更大的crop size提高了所有指标,蛋白-蛋白界面提升最高(下面的拓展图2)。
拓展图2|初始训练是蓝色曲线,微调阶段1橙色曲线,绿色曲线是微调阶段2。红色虚线/紫色虚线分别为90%/97%性能时的训练步。
4模型表现
如图3所示为AF3所进行的一系列结构预测示例,AF3可以根据输入的氨基酸序列、残基修饰、配体SMILES进行结构预测。
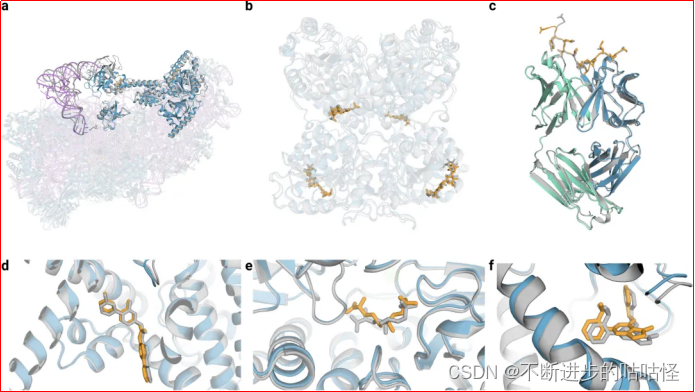
图3|AF3的结构预测示例。
a,7663个残基的40S小核糖体亚基蛋白、不透明蓝色的翻译始因子eIF1A和eIF5B、紫色为18S核糖体RNA、不透明紫色的Met-tRNA,复合物置信度LDDT=87.7。
b,糖基化的EXTL3二聚体,与共晶RMSD=1.1Å。
c,间皮素C端肽,与单克隆抗体15B6 结合,DockQ为0.85。
d,临床阶段抑制剂LGK974与带有WNT3A肽的PORCN蛋白结合形成复合物,小分子RMSD=1Å。
e,(5S,6S)-O7-sulfo DADH与AziU3/U2的复合体,与共晶RMSD=1.92Å。
f,NIH-12848类似物与PI5P4Kγ变构位点结合,与共晶RMSD=0.37Å。
4.1蛋白质-小分子配体结构预测
蛋白质-配体结构预测的AF3性能在PoseBusters数据集上进行了160项评估,该数据集由 428个蛋白质-配体结构组成,其中161个在2021年以后加入到PDB。于是在更早的训练集训练了单独的AF3版本(保证无数据泄露)。下图成功率指配体RMSD小于 2Å 的百分比。从下图可见,AF3大大优于基于结构的Vina,以及RFAA模型。

图1c, 蛋白-小分子 |在PoseBusters数据集上AF3的成功率。纵坐标成功率定义为配体RMSD < 2 Å 的百分比,N表示靶点的数量。
4.2蛋白质-核酸符合物或RNA单体
AF3够比RoseTTAFold2NA更准确地预测蛋白质-核酸复合物和 RNA 结构(下面的图1c)。我们没有与RFAA比较,因为RFAA精度低于RoseTTAFold2NA。从下图可见,AF3的蛋白-核酸对接成功率远远高于RoseTTAFold2NA算法。
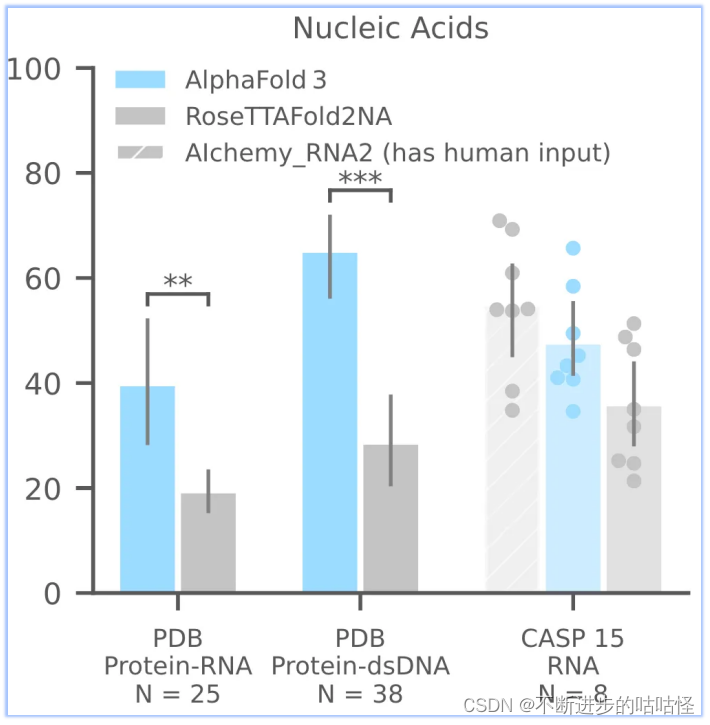
图1c,核酸相关相互作用 |PDB数据库中蛋白-RNA,蛋白-双链DNA;CSAP15比赛中RNA单体的成功率。成功率针对复合物是iterface LDDT,单体RNA仅为LDDT。N表示靶点的数量。
此外还评估了CASP15的10个RNA单体,与RoseTTAFold2NA和AIchemy_RNA2对比,AIchemy_RNA2表现出更加优异的性能(上图c,右边),详细结果在拓展图5a。

拓展图5a|在CSAP 15 RNA数据集上,AF3和RoseTTAFold2NA和AIchemy_RNA2对比,横坐标是10个RNA编号。纵坐标分别为LDDT、TM score、GDT等置信度指标。
AF3没有达到人类专家辅助的 AIchemy_RNA2的性能 (上图)。单独预测核酸DNA/RNA(没有蛋白质)的准确性LDDT的进一步分析显示在扩展图5b中。
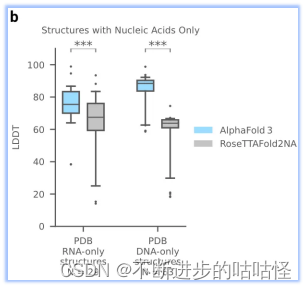
扩展图5b|在单独核酸DNA/RNA上评估LDDT,AF3与RoseTTAFold2NA对比。

扩展图5c|蛋白-dsDNA复合物示例,左图以pLDDT绘图,右图以chain绘图
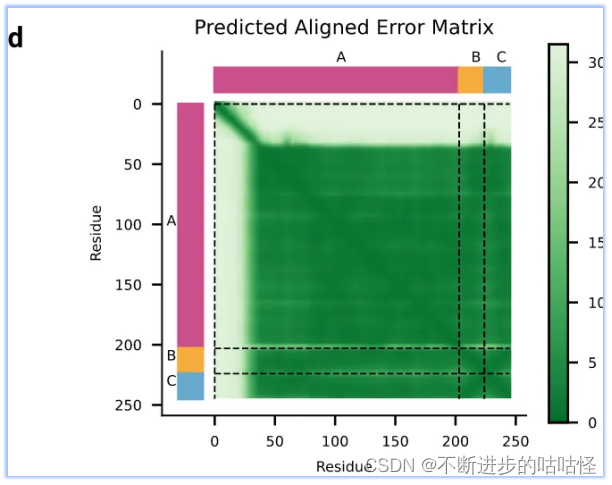
拓展图d|预测蛋白-核酸结构的PAE误差,不同颜色代表不同链A/B/C。
4.3共价修饰
AF3还可以准确预测共价修饰(键合配体、糖基化、修饰的蛋白残基和核酸碱基)(下图 1c)。修饰包括对任何聚合物残基(蛋白质、RNA或DNA)的修饰。成功率定义为RMSD < 2 Å。

图1c,共价修饰|纵坐标成功率定义为配体RMSD < 2 Å 的百分比,N表示靶点的数量。依次为,蛋白键和小分子、蛋白糖基化、蛋白修饰、DNA/RNA修饰。
扩展图6显示了具有共价修饰的蛋白质、DNA和RNA预测结构的示例,包括分析磷酸化对预测的影响。

拓展图6a|在磷酸化 (SEP、TPO、PTR、NEP、HIP)场景,AF3的预测成功率,PTM代表转录后修饰。
4.4蛋白-蛋白/蛋白质单体
在扩展建模能力的同时,AF3相对于 AlphaFold-Multimer v2.3也提高了蛋白质复合物准确度,结果见下图c。抗体蛋白质相互作用预测尤其显示出显着的改善。蛋白质单体LDDT的改善也有。

图1c, 蛋白相关相互作用或单体蛋白 |蛋白Multimer、蛋白-抗体、蛋白单体在Recent PDB评估集上的预测成功率。蛋白Multimer、蛋白-抗体的成功率定义为DockQ > 0.23。蛋白单体使用LDDT指标定义成功率。N表示靶点的数量。
5总结
****分子生物学的核心挑战是理解并最终调节生物系统的复杂原子相互作用。****AlphaFold3作为一个可以预测所有生物大分子的通用模型,表明可以在统一的框架中准确预测各种生物分子系统的结构,该系统对所有所有分子交互类型具有很强的覆盖率和泛化率。但同样也有局限性:
- ****立体化学的局限性。****手性问题:即使输入了正确的手性参考结构,AlphaFold3有时仍会输出违反手性的模型。原子重叠:在某些情况下,如蛋白-核酸复合物,模型可能会产生原子重叠的现象。
- ****幻想问题。****从非生成式模型AF2到基于扩散的AlphaFold3的转变引入了无序区域的虚假结构。这些区域虽然被预测为低置信度,但缺乏AF2模型在无序区域产生的独特带状外观。为了解决这个问题,AlphaFold3采用了AF2的预测结果进行蒸馏训练,并引入了鼓励更大溶剂接触表面积的惩罚。
- ****动力学问题。****AlphaFold3主要预测静态结构,而无法捕捉生物分子系统在溶液中的动态行为。
- 特定目标的精度问题 ****。****在某些情况下,如E3泛素连接酶,AlphaFold3可能无法准确模拟其在不同状态下的构象。例如,它可能只预测出与配体结合时的封闭状态,而忽略了在载脂蛋白状态下的开放构象。
- 准确性与计算成本 ****。****为了提高预测的准确性,可能需要生成和评估大量的模型结果,这将导致显著增加的计算成本。特别是在抗体-抗原复合物的预测中,随着模型随机种子数量的增加,预测效果有所提升,但这也意味着需要更多的计算资源。
AF3的性能表明,开发正确的深度学习框架可以大大减少获得这些任务的生物学相关性能所需的数据量,并放大已经收集的数据的影响。下一步作者将进一步改进结构建模,采用置信度更高的实验方法以提高模型的泛化能力。
文献信息:
Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630 , 493--500 (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3 | Nature
另外参考: