零基础入门转录组数据分析------机器学习算法之SVM-RFE(筛选特征基因)
目录
- 零基础入门转录组数据分析------机器学习算法之SVM-RFE(筛选特征基因)
-
- [1. SVM-RFE基础知识](#1. SVM-RFE基础知识)
- [2. SVM-RFE(Rstudio)------代码实操](#2. SVM-RFE(Rstudio)——代码实操)
-
- [2. 1 数据处理](#2. 1 数据处理)
- [2. 2 构建SVM-RFE模型](#2. 2 构建SVM-RFE模型)
- [2. 3 SVM-RFE结果简单可视化](#2. 3 SVM-RFE结果简单可视化)
- 注:配套资源只要改个路径就能运行,本人已检测过可以跑通,请放心食用,食用过程遇到问题,可先自行百度,实在解决不了可以私信
您首先需要了解本贴是完全免费按实际案例分享基础知识和全部代码,希望能帮助到初学的各位更快入门,但是尊重创作和知识才会有不断高质量的内容输出 ,如果阅读到最后觉得本贴确实对自己有帮助,希望广大学习者能够花点自己的小钱支持一下作者创作(条件允许的话一杯奶茶钱即可),感谢大家的支持~~~~~~ ^_^ !!!
注:当然这个并不是强制的哦,大家也可以白嫖~~,只是一点点小的期盼!!!
祝大家能够开心学习,轻松学习,在学习的路上少一些坎坷~~~
1. SVM-RFE基础知识
1.1 SVM是什么?
SVM(Support Vector Machine)算法是一种常见的监督学习算法,用于进行二分类或多分类任务。其主要思想是找到一个最优的组合,将不同类别的样本分隔开
1.2 RFE是什么?
RFE(Recursive Feature Elimination)算法是一种常用的特征选择方法,其通过逐步迭代,训练模型并剔除最不重要的特征,然后再次训练模型,直到达到指定的特征数量或达到某个性能指标。
1.3 SVM-RFE是什么?
SVM-RFE算法是SVM和RFE算法的结合,通过SVM模型进行特征的重要性评估,并利用RFE的迭代过程逐步剔除不重要的特征。
1.4 SVM-RFE原理?
SVM-RFE算法基于SVM的最大间隔原理,通过模型训练样本,对每个特征进行得分排序,之后利用RFE算法逐步迭代的方式:去掉最小特征得分的特征,然后用剩余的特征再次训练模型,进行下一次迭代,最后选出最优的特征组合。
1.5 SVM-RFE的优势?
- 结合两种算法的优点: SVM算法主要关注于分类任务,通过寻找最优组合来实现不同类别样本的分离,但它本身并不具备特征选择的能力,而RFE算法则会迭代剔除不重要的特征,这样就能够在模型训练的同时进行特征选择,从而选出对分类结果最有影响的特征子集
- 考虑特征之间的关联: 在每一轮迭代中,都会重新计算特征的权重,这样可以更好地考虑特征之间的关联关系,避免特征选择过程中的信息丢失。
- 提高模型泛化能力: 通过剔除不重要的特征,SVM-RFE算法能够减少特征空间的维度,降低模型的复杂性,从而提高模型的泛化能力。
-提高模型可解释能力: 筛选出的特征子集往往具有更好的可解释性,因为它们是数据中最具代表性的特征组合,同时算法会自动计算特征重要性并进行迭代,这样就减少了人为选择变量的可能性。
1.6 SVM-RFE的本质是什么?
筛选出一些关键特征,这些关键特征相对于其他特征来说,区分样本的能力更加精确。
举个栗子: 输入了8个基因的表达矩阵,此时基于这8个基因构建模型去观测模型对与样本的区分能力,发现区分准确性为80%,之后通过迭代的方式逐一剔除相对不重要的基因,最后发现剔除3个"不重要"基因后剩余5个基因构建的模型区分样本的准确性为95%(最高),那么此时认为这5个基因组合而成的模型为最优模型,这5个基因就作为对分类结果最有影响的特征子集被输出了。
综上所述: SVM-RFE就是一种用来筛选关键特征的方法(其结合了SVM算法的优点,同时也结合了RFE算法的优点),这个关键特征可以是临床指标,也可以是重要基因等,并且在关键特征选择的时候避免了人为的选择(算法自动迭代),输出基因重要性提高了可解释性。
2. SVM-RFE(Rstudio)------代码实操
本项目以TCGA------肺腺癌为例展开分析
物种:人类(Homo sapiens)
R版本:4.2.2
R包:tidyverse,caret,ggplot2,cowplot,ggplotify废话不多说,代码如下:
2. 1 数据处理
设置工作空间:
r
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./09_SVM-RFE')){
dir.create('./09_SVM-RFE')
}
setwd('./09_SVM-RFE/') 加载包:
r
library(tidyverse)
library(caret)
library(ggplot2)
library(cowplot)
library(ggplotify)导入要分析的表达矩阵train_data,并对train_data的列名进行处理(这是因为在读入的时候系统会默认把样本id中的"-"替换成".",所以要给替换回去)
r
train_data <- read.csv("./data_fpkm.csv", row.names = 1, check.names = F) # 行名为全部基因名,每列为样本名
colnames(train_data) <- gsub('.', '-', colnames(train_data), fixed = T)train_data如下图所示,行为基因名(symbol),列为样本名

导入分组信息表group
r
group <- read.csv("./data_group.csv", row.names = 1) # 为每个样本的分组信息(tumor和normal)
colnames(group) <- c('sample', 'group')group如下图所示,第一列sample为样本名,第二列为样本对应的分组 (分组为二分类变量:disease和control)

导入要筛选的基因hub_gene(8个基因)
r
hub_gene <- data.frame(symbol = gene <- c('ADAMTS2', 'ADAMTS4', 'AGRN', 'COL5A1', 'CTSB', 'FMOD', 'LAMB3', 'LAMB4'))
colnames(hub_gene) <- "symbol"hub_gene如下图所示,只有一列:8个基因的基因名

从全部的基因表达矩阵中取出这8个基因对应的表达矩阵,并且与之前准备的分组信息表进行合并
r
dat <- train_data[rownames(train_data) %in% hub_gene$symbol, ] %>%
t() %>%
as.data.frame() # 整理后行为样本名,列为基因名
dat$sample <- rownames(dat)
dat <- merge(dat, group, var = "sample")
dat <- column_to_rownames(dat, var = "sample") %>% as.data.frame()
table(dat$group)
dat$group <- factor(dat$group, levels = c('disease', 'control'))dat如下图所示,行为基因名,前8列为基因对应的表达矩阵,第9列为合并的分组信息表

2. 2 构建SVM-RFE模型
构建SVM-RFE模型:
(1)使用rfeControl函数来设置递归特征消除过程中的交叉验证(CV)参数。这里指定了使用caretFuncs(一系列预定义的训练和预测函数,包括错误率等评估指标)作为评估函数,使用cv(交叉验证)作为方法,并设置交叉验证的次数为10次(number = 10)。
(2)执行递归特征消除 (rfe)算法构建模型rfe函数常用参数介绍如下:
- x参数------是要输入的基因表达矩阵(也称为特征或自变量)
- y 参数------这是要区分的目标变量。这里指向的是dat$group,根据前面得知这里是二分类变量分组------disease和control
- sizes 参数------这个参数指定了特征子集(要分析的范围),这里用c(1 : num)表示从1个特征到所有特征(除了最后一列)的所有可能组合都将被评估。
- rfeControl参数------这个参数传递了之前定义的交叉验证控制参数。
- method 参数------这个参数指定了用于评估特征子集性能的机器学习算法,这里是使用svmRadial方法------是使用支持向量机作为底层分类器,并且该SVM使用的是径向基函数核 (关于这个方法的介绍在这里不做展开介绍,感兴趣的小伙伴可以自行检索下)
(构建SVM-RFE模型中比较关注的参数就是上述的这些,当然还有其他参数,如果想深入了解可自行查看官方说明文档)
r
set.seed(21) # 设置种子
control <- rfeControl(functions = caretFuncs, method = "cv", number = 10) # cv 交叉验证次数10
# 执行SVM-RFE算法
num <- ncol(dat)-1
results <- rfe(x = dat[, 1:num], # 除去最后一列,其余列均为预测变量(也就是hubgene的表达量)
y = dat$group, # 分组信息
sizes = c(1:num),
rfeControl = control,
method = "svmRadial"
)注:在构建模型的时候切记要设置种子(设置随机种子是为了确保结果的可重复性。由于交叉验证涉及随机分割数据,因此设置种子可以确保每次运行代码时,数据的分割方式都是相同的,从而得到相同的模型结果)
注2:这一步在构建模型的时候花费的时间会比较长,属于正常情况!
接下来从构建的最优模型中提取出最优模型组合,并保存关键基因
r
## 结果分析
svmrfe_result <- data.frame(symbol = predictors(results)) ## 7个基因
write.csv(svmrfe_result, file = 'svm_rfe_gene.csv')svmrfe_result中就对应着最优模型组合中的基因,如下图所示,可以看到只有7个基因,有个基因被剔除了,这7个基因就被认为是更加重要的特征基因。

2. 3 SVM-RFE结果简单可视化
接下来一步就是要对SVM-RFE结果进行简单可视化,毕竟文章里是要放图的,并且图片展现的效果会更好!!!
r
# SVM-RFE结果简单可视化
p1 <- plot(results, type=c("o"),
xgap.axis = 1)
p1 <- as.ggplot(plot_grid(p1))+
labs(title="SVM_RFE_analyse", x="", y = "",size=25) +
# theme_bw()+
theme(plot.title = element_text(hjust =0.5,colour="black",face="bold",size=25),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
legend.text = element_blank(),
legend.title = element_blank(),
legend.position = "none",
panel.grid.major = element_blank(),
panel.grid.minor = element_blank())
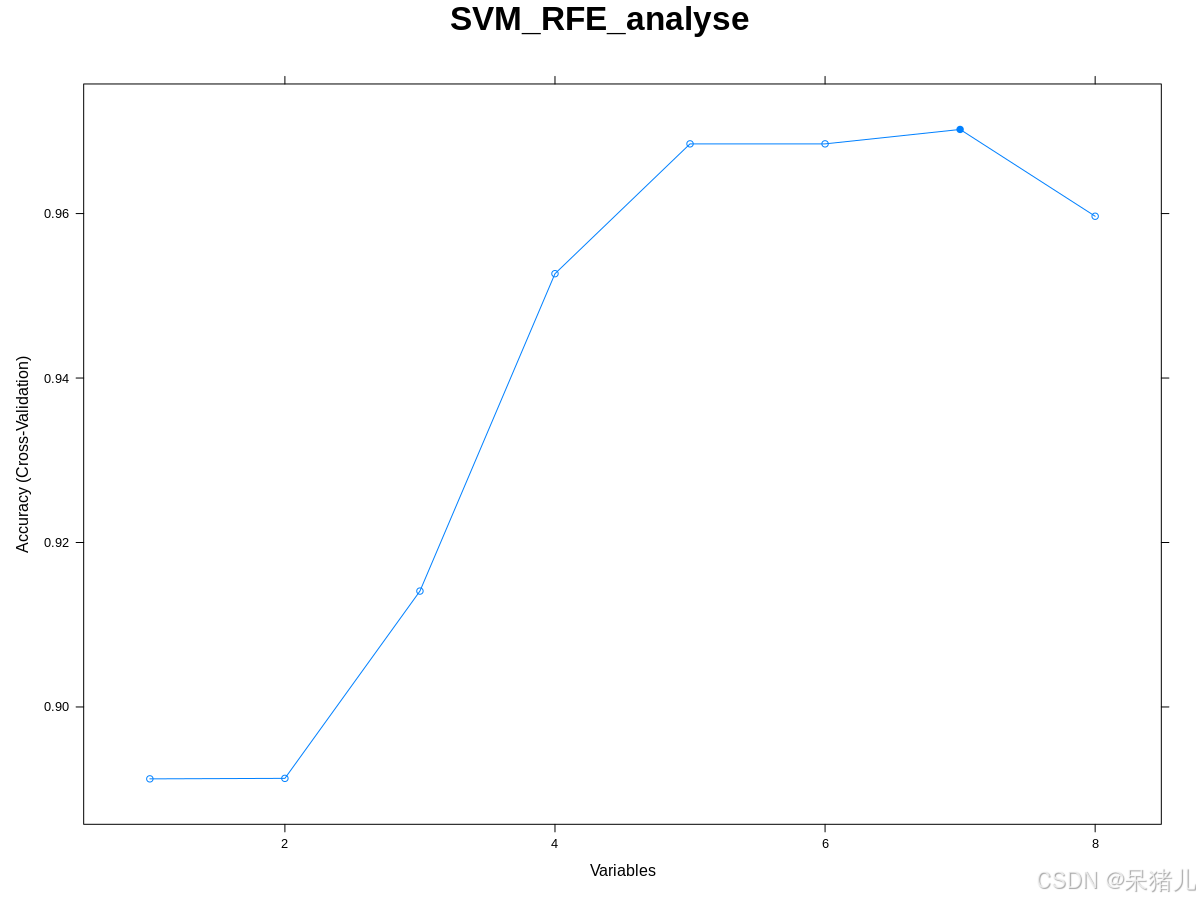
p1SVM-RFE结果如下图所示,横坐标为变量对应的数目(这里指的是基因数目),纵坐标为区分样本的准确性,可以看到当所有基因都存在的时候(最右侧的点)区分准确度为0.96,剔除掉一个相对不重要的基因后区分准确度最高,之后再剔除基因就会导致模型区分准确度下降。
结语:
以上就是SVM-RFE算法筛选关键基因的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,点赞关注不迷路!!!
与教程配套的原始数据+代码+处理好的数据见配套资源
注:配套资源只要改个路径就能运行,本人已检测过可以跑通,请放心食用,食用过程遇到问题,可先自行百度,实在解决不了可以私信
- 目录部分跳转链接:零基础入门生信数据分析------导读