摘要
设计兼具高稳定性和高活性的蛋白质突变体是蛋白质工程中关键而富有挑战的任务。本文提出的深度学习模型PRIME无需特定蛋白的预实验突变数据,即可设计出稳定性与活性提升的蛋白质突变体。基于温度感知语言建模技术,PRIME在涵盖283套蛋白质检测实验的公共突变数据集上展现出超越现有最优模型的预测能力。通过对五种蛋白质的验证,我们评估了PRIME推荐的30至45个单点突变对多种蛋白特性的影响,包括热稳定性、抗原-抗体结合亲和力、非天然核酸聚合能力及极端碱性耐受性。所有测试蛋白质中,超过30%的PRIME推荐突变体在目标特性上显著优于突变前蛋白。我们基于PRIME开发了可快速获得活性与稳定性增强的多位点突变体的高效方法。由此证明PRIME在蛋白质工程领域具有通用性。
引言
蛋白质是生命系统的基础组分,在酶催化、细胞代谢、免疫应答、信号转导和物质运输等众多生物过程中发挥关键作用。其生物学价值之外,蛋白质对医疗(治疗剂与靶点)、食品(加工与保鲜)、酿造(生产关键)及化工(反应核心催化剂)等工业领域至关重要。此外,蛋白质构成体外诊断检测的基石,广泛应用于疾病检测与监测。然而,天然生物体提取的"野生型"蛋白质常需改造以适应工业应用场景。工业环境所需的理化条件(如温度)常与天然生物环境存在巨大差异。因此,需通过突变工程改造蛋白质,提升其理化特性以满足多样化应用需求。此类改造旨在提升极端温度或pH条件下的稳定性,或增强酶活性与特异性。面向工业应用的蛋白质优化需经历突变-筛选-选择的迭代循环,属于劳动密集型且耗时的过程。
随着计算模拟及相关技术的发展,Rosetta、ABACUS、FoldX等基于物理或统计势能函数的蛋白质热稳定性优化软件相继涌现。尽管这些计算方法能提供较准确的稳定性预测,但对蛋白质生物活性的预测能力有限。改造蛋白质生物活性通常需耗费数年时间深入研究其作用机制,这是理性蛋白质设计的主要途径。然而这种机制研究耗时耗力,难以满足日常工业用关键酶类的改造需求。
近年来深度学习在蛋白质工程中广泛应用。大规模蛋白质语言模型(PLMs)通过自监督学习理解序列语义与语法,在蛋白质适应性预测中展现显著效果,甚至可实现零样本预测。零样本预测指模型无需预实验突变数据即可推荐提升蛋白质性能的突变位点。但多数预训练PLMs对蛋白质稳定性(工程关键指标)的预测精度仍显不足。监督学习方法虽预测精度高,却需依赖高通量实验获取数百至数千数据点。因资源限制,该方法对多数蛋白质不具普适性。
本研究采用含9600万组宿主菌株最适生长温度(OGT)的数据集。宿主菌最适生长温度(OGT)被证实与蛋白最适酶活温度、熔解温度等参数强相关。基于此,我们开发了深度学习模型PRIME(智能掩蔽预训练与环境温度预测蛋白质语言模型,Protein language model for Intelligent Masked pretraining and Environment)。预训练中PRIME采用基于Transformer模型的掩蔽语言建模(MLM)任务。该任务通过氨基酸自然概率分布掩蔽蛋白质序列,再训练模型重建原序列。此过程使PRIME掌握蛋白质序列的语义与语法特征。同时通过多任务学习捕获序列的温度关联特性。该机制使PRIME优先为具温度耐受性且符合自然生物学规律的序列赋高分。PRIME以实现广谱细菌菌株最适生长温度(OGT)预测为目标进行训练。因此其高分序列天然关联到极端温度等严苛环境下的强韧性与存活率。这使PRIME特别适用于需高温耐受性的工业酶与蛋白质的优化设计。在预测热稳定性熔解温度(Tm)变化与突变序列适应性方面,PRIME展现出显著优于现有尖端模型的性能。
为验证模型效能,我们选取五种不同蛋白质开展湿实验验证:涵盖LbCas12a、T7RNA聚合酶、肌酸酶、非天然核酸聚合酶及生长激素纳米抗体重链可变区(VHH)。在零预实验突变数据条件下,PRIME筛选出多组高评分单点突变体进行实验测试。结果显示:超30%突变体的理化性质显著提升,包括热稳定性、催化活性、抗原抗体结合亲和力,以及聚合非天然核酸、耐受极端碱性等特殊功能。
面向医药与工业应用的蛋白质工程面临两大挑战:一是筛选有益单点突变位点,二是构建多位点叠加的深度突变体。后者尤为困难------实验常观察到两个阳性单点突变组合后,双位点突变体性能反低于单点突变体。如绿色荧光蛋白高通量筛选所示:阴性上位效应(双突变体荧光强度低于两单点突变适应度线性叠加值)的发生概率超阳性效应100倍。
基于PRIME框架,我们开发了多突变位点叠加策略。例如针对T7RNA聚合酶,经三轮AI-实验迭代(总测试突变体<100个),成功获得含12处突变的突变体,其热稳定性超越行业龙头New England Biolabs的商用热稳定版本。在含多结构域(1228个氨基酸)的LbCas12a蛋白中应用相同策略:经三轮AI-实验迭代(总测试突变体<100个),获得迄今热稳定性最佳的8点位突变体------Tm值较野生型提高6.5°C,且在目标条件下保持相当或更强的反式切割活性。
进一步在T7 RNA聚合酶与LbCas12a案例中发现:PRIME能自动整合不同功能域的负性单点突变,构建成多位点深度突变体以提升其适应性。该发现为蛋白质工程师开辟了新路径------利用负突变提升蛋白质适应性。此类负突变发生频率远高于正突变,却在传统蛋白质工程中被预先排除。
结果
PRIME架构
PRIME是基于Transformer架构的预训练模型(图1A)。包含三个核心组件:
-
序列特征提取编码器------Transformer编码器模块,用于捕获序列潜在特征表示;
-
掩蔽语言建模(MLM)模块------引导编码器学习氨基酸的上下文表征;该模块同时用于突变体评分;
-
最适生长温度(OGT)预测模块------基于潜在特征预测蛋白质宿主生物体的OGT。
模型参数与训练细节详见方法部分。
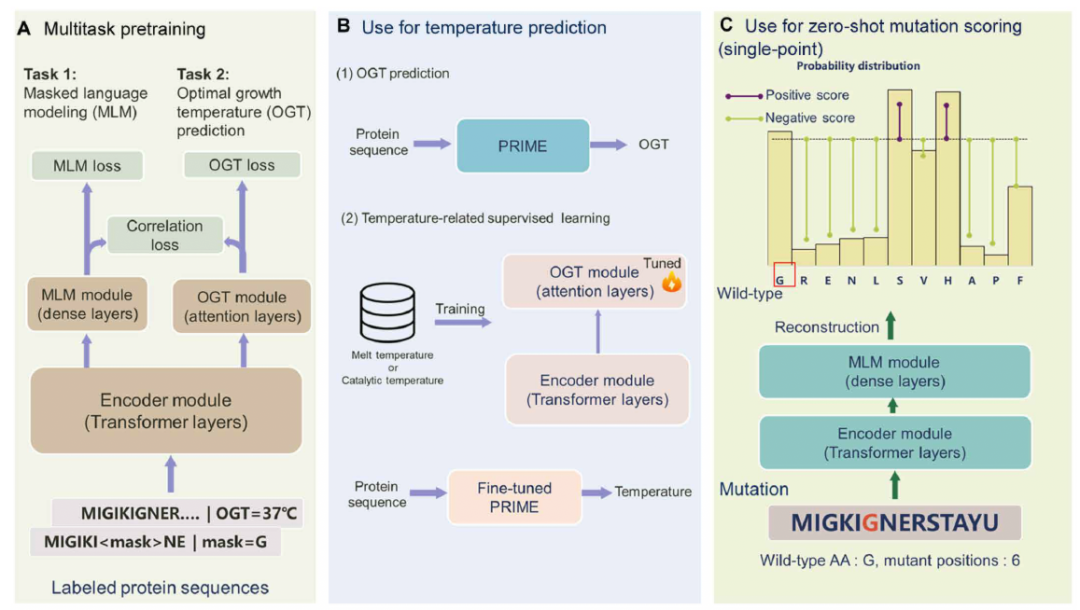
图1. PRIME架构及应用概览。(A)PRIME架构设计。采用基于BERT的Transformer编码器,集成两个专用模块:MLM任务模块与OGT预测任务模块。训练目标包含三种损失函数:基于交叉熵的MLM损失、均方误差准则的OGT损失、逆皮尔逊相关系数的相关性损失。(B)PRIME温度预测应用。可预测蛋白序列OGT,并能通过其他温度数据集(如熔解温度或最适催化温度)进行微调。(C)PRIME单点突变评分应用。通过MLM模块重建野生型序列,生成突变位点氨基酸概率分布。通过突变型与野生型氨基酸的对数似然比量化突变效应。(D)多位点突变体生成策略。第一步:评估单点突变效应,筛选Top-K突变体进行实验适应性检测;第二步:实验数据用于PRIME模型微调,微调后模型预测多位点突变体适应性;第三步:根据预测结果筛选Top-K多位点突变体开展进阶实验。
PRIME预训练目标
包含三个训练目标:掩蔽语言建模目标、最适生长温度预测目标与相关性目标,具体描述如下:
掩蔽语言建模:MLM作为序列数据表征的预训练方法------ 以含噪蛋白质序列为输入,随机将序列标记掩蔽为"
最适生长温度预测。该训练目标在监督条件下优化:基于含9600万条OGT标注的蛋白质序列数据集训练模型;输入为蛋白质序列,OGT模块输出0°C-100°C区间的温度值;关键点在于OGT与MLM模块共享编码器------此架构使模型同步捕获氨基酸上下文信息与温度相关序列特征(图1B)。
相关性目标。该学习目标旨在关联两个指标:通过OGT预测值反馈调节MLM评分。处理单点突变序列组时,OGT模块输出其温度值,MLM模块输出突变评分;通过最大化突变评分与预测OGT值的皮尔逊相关系数,实现突变OGT值与评分的协同优化。目标函数设定为皮尔逊相关系数最大化。之所以选用皮尔逊相关系数是因为其具有反向传播所需的可微分特性,而非不可微分的斯皮尔曼相关系数。
实验中尝试采用均方误差(MSE)损失函数对齐MLM与OGT预测值(表S1),发现其性能逊于皮尔逊相关损失函数。因MSE损失仅针对单序列对齐MLM与OGT值,导致个体数据损失波动显著,且MLM评分绝对值对研究意义有限;而相关性损失基于突变序列集计算,更契合蛋白质工程中评估突变数据集的实际应用场景。
零样本单点突变评分
基于MLM训练的模型能依据上下文环境输出特定位点氨基酸的出现概率;据此进行单点突变评分:以野生型蛋白的氨基酸为参照,对比其与突变型氨基酸的出现概率;基于突变位点处的对数似然比量化突变评分(图1C,方法详见原文)。
基于同源序列的MLM微调增强PRIME单点突变预测性能
尽管PRIME在零样本突变效应预测中表现优异,我们发现对目标蛋白同源序列进行语言建模模块的无监督微调可进一步提升效果,且无需引入实验数据监督。具体操作中:采用目标蛋白同源序列作为无监督数据集,对PRIME及ESM2-650M的编码器与MLM模块联合优化;评估结果证实该方法能提升PRIME与ESM-2的突变效应预测精度。
PRIME在突变蛋白序列适应性预测中超越现有最优方法
本研究将PRIME与当前主流模型进行零样本热稳定性预测能力对比,涵盖深度学习模型ESM-1v、ESM-2、MSA-transformer、Tranception-EVE、CARP、MIF-ST、SaProt、Stability Oracle,以及传统计算方法GEMME和Rosetta。需指出:MIF-ST、SaProt与Rosetta整合了蛋白质结构信息,其余模型仅依赖序列信息。分析采用MPTherm、FireProtDB及ProThermDB数据集,包含66项检测数据:所有单点突变均记录熔解温度变化值(ΔTm),实验在相同pH环境下完成,每种蛋白至少包含10个数据点。同时整合深度突变扫描(DMS)数据,尤其来自ProteinGym数据库的检测结果。
ProteinGym数据库构建了精细的替代基准,其内容特征是对约250万个错义变体进行实验表征与评估。这些变体分布于217项独立DMS (deep mutational scanning)检测中,涵盖酶催化活性、结合亲和力、稳定性及荧光强度等多元蛋白质特性。ProteinGym替代基准中错义变体的全面汇编构建了稳健而详实的资源库,为深入解析海量错义变体的复杂特性提供支撑。该资源库由此成为系统研究蛋白质突变多样性及其关联特性不可或缺的宝贵资源。
这些综合性数据集支持系统研究特定突变对蛋白适应性与热稳定性的影响,为PRIME等先进预测模型的开发验证提供了基础。对比实验揭示不同建模方法的相对性能,凸显PRIME在零样本蛋白质突变预测中的潜力。结果展示于图2A与表S2。可见PRIME在蛋白适应性与稳定性预测中全面优于其他方法。
-
ProteinGym基准测试中,PRIME以0.486分(对比SaProt模型0.457分,P=1×10^−4, Wilcoxon检验)领先。
-
ΔTm数据集评估显示:PRIME得分0.437显著优于Stability Oracle的0.412(P=9×10^−3, Wilcoxon检验)。
-
在ProteinGym稳定性子集(ProteinGym-stability)中对比:PRIME仍全面超越其他模型。
需强调:PRIME所用的OGT并非蛋白Tm直接表征,二者存在相关性。
部分源自嗜热菌的酶实际热稳定性不高。但即便使用精度稍逊的OGT替代Tm,PRIME仍显著优于未整合OGT的模型。如同架构的ESM-2在ΔTm数据集仅得0.330分。我们认为获取大量天然蛋白精确Tm值将显著提升PRIME性能。上述发现印证PRIME在提升蛋白质热稳定性等适应特性工程中的潜力。PRIME全面超越传统计算方法与其他深度学习模型,彰显其卓越效能。
近期诸如SaProt等将蛋白质结构信息整合到PLMs中的研究提升了稳定性预测能力;但SaProt及MIF-ST、Stability Oracle等结构模型需输入蛋白质结构数据------这类数据本身存在噪音,且受限于湿实验解析或AlphaFold预测等高精度结构的可获取性;导致其应用存在局限性。仅需序列输入的PRIME在以完整版ProteinGym(含217套数据集)为基准测试时,已超越当前最优模型SaProt。作为纯序列模型,PRIME相较ESM-2等其他PLMs不仅显著提升稳定性数据集预测能力,更在非稳定性数据集(尤其涉及活性的数据集,见表S2)中实现更优性能。
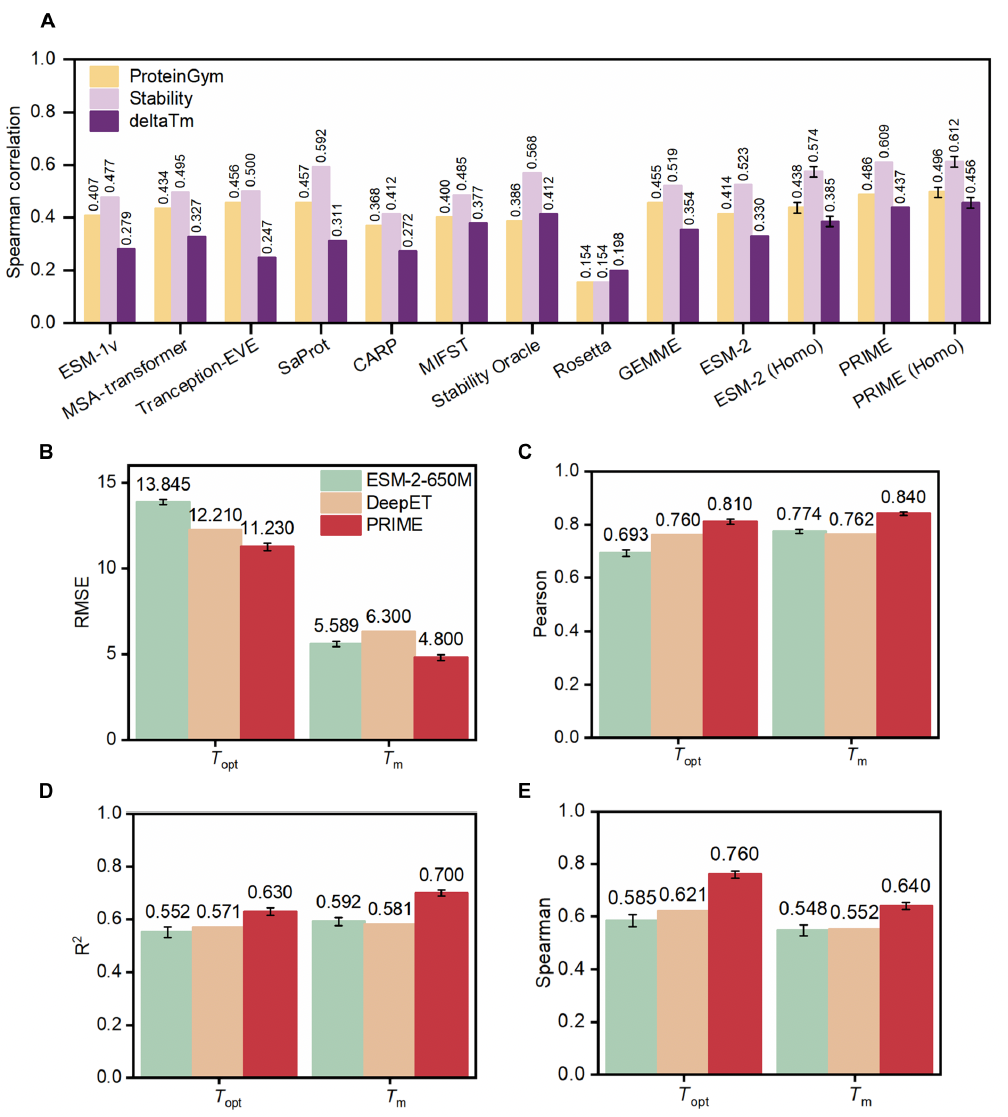
图2. PRIME与其他方法性能对比。(A)ΔTm与ProteinGym数据集的无监督模型基准测试。"PRIME(同源序列)"指基于ProteinGym或ΔTm数据集中目标蛋白同源序列,采用MLM损失函数对PRIME模型进行微调。(B-E)熔解温度(Tm)与最适酶活温度(Topt)的监督预测。监督测试中,PRIME与ESM-2采用三种随机种子训练,DeepET结果引自文献(82)。模型精度与预测能力评估指标:RMSE(均方根误差)(B)、皮尔逊相关性(C)、R2(决定系数)(D)、斯皮尔曼相关性(E)Tm与Topt数据集划分参照文献(82)。野生型蛋白结构取自PDB数据库,PDB缺失结构用AlphaFold2建模后输入Rosetta与MIF-ST。图2相关数据点及P值检验详见附表S2与S3。
除零样本任务外,我们验证了PRIME的表征能力与迁移性能:通过对两项温度相关下游任务进行全局微调的监督训练实现(图1B);因PRIME预训练包含宿主菌最适生长温度信息,预计其在其他温度关联蛋白特性预测中同样具优势;图2(B-E)与表S3显示:在预测天然蛋白质熔解温度(Tm)及最适酶活温度(Topt)任务中,PRIME仍优于其他监督方法;鉴于Tm与Topt对蛋白质的重要性,PRIME仅凭序列输入即可快速批量标注天然蛋白热学特性的能力,对实际应用中的天然蛋白注释工程具有显著价值。
我们深入解析了PRIME三大核心组件(OGT预测模块、MLM模块及相关性目标)的独立贡献。如表S1显示:仅依赖单一模块会降低模型性能;其中MLM模块在所有零样本基准测试中影响最为显著;OGT模块在ΔTm预测中具关键作用------完整PRIME得分为0.437,而缺失OGT模块的PRIME/-OGT仅得0.362(P=3×10−2, Wilcoxon检验);相关性目标同样显著影响ΔTm预测:缺失该模块的PRIME/-correlation得分为0.429(P=4×10−2, Wilcoxon检验);
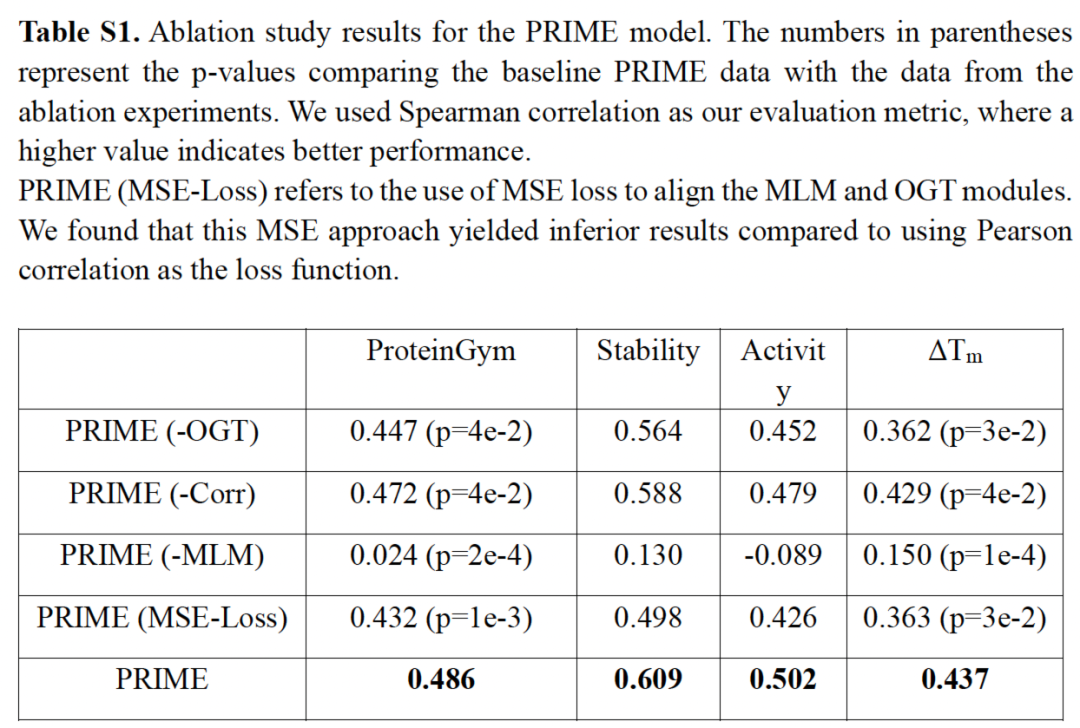
在ProteinGym基准测试中,OGT与相关性目标仍保持显著影响。该结果印证三大组件协同工作对PRIME实现最优性能的必要性;其协同效应使模型更精准捕捉蛋白质序列与热稳定性间的复杂关系,从而提升预测能力;组件集成确保对蛋白质序列信息的全面理解,这使PRIME在性能上持续超越其他尖端模型。
我们进一步评估了PRIME在其他监督式蛋白质工程任务中的表现:在包含12项任务的蛋白质适应性景观推断(FLIP)基准测试中,PRIME全面领先ESM-1b、ESM-1v、ESM-2和CARP模型,展现出优异的外推能力------尤其对高复杂度突变效应的预测(表S4)。需特别指出:FLIP中AAV与GB1两项任务要求基于单点突变适应性数据预测多位点深度突变体适应性,这对筛选蛋白质工程终产物至关重要。该能力源于预训练中标记级MLM与序列级OGT特征的对齐------使模型同步掌握天然序列热学特性与突变序列热稳定性排序规律。由于蛋白质热稳定性、结合亲和力等极端环境耐受性均遵循结构稳定性物理原理,PRIME在FLIP基准的天然蛋白热稳定性(Meltome)及突变蛋白结合亲和力(AAV/GB1)预测任务中展现出卓越外推能力。此即PRIME在该类任务中优于ESM系列的原因。在FLIP的Meltome数据集任务(预测人源蛋白Tm)中,整合OGT信息的PRIME持续超越同架构模型ESM-2;这表明尽管预训练仅基于细菌源蛋白OGT数据,PRIME仍能精准预测跨物种蛋白的Tm属性。由此印证其强大的通用能力。
PRIME设计蛋白单点突变体的湿实验验证:多蛋白多场景工程测试
蛋白质工程实践主要采用两步策略:筛选提升活性或稳定性的正向单点突变,再通过贪婪搜索法组合成具有预期目标性能的多位点突变体;因此阳性单点突变的精准识别是蛋白质工程成功的核心前提。
为验证该方法效力,选取五种蛋白开展单点突变设计测试:LbCas12a、T7RNA聚合酶、肌酸酶、非天然核酸聚合酶(Tgo-D4K)及生长激素纳米抗体重链可变区(VHH)。基于Uniclust30数据库,为各目标蛋白选取3万条同源序列微调PRIME;每个蛋白采用五种随机种子独立微调;综合五种模型预测结果取平均值,生成各蛋白单点饱和突变评分表;基于突变序列相对野生型的似然比(参见突变蛋白序列评分策略),PRIME对所有单点突变体排序;筛选催化活性位点或结合口袋6埃范围外的Top30-45突变体进行实验;因活性位点的突变可能颠覆蛋白功能,本研究采用保守策略避免催化能力受损;值得注意的是:PRIME推荐突变的实际效应需实验确认;各蛋白存在差异化改造需求: 具体目标为------LbCas12a、T7 RNA聚合酶及肌酸酶侧重提升热稳定性;非天然核酸聚合酶(Tgo-D4K)旨在加速2'-氟阿拉伯糖核酸(FANA)聚合速率;VHH则需增强强碱性环境(pH>13)稳定性。完整实验结果详见后续章节。
PRIME可依据活性与稳定性对单点突变体进行排序;但PRIME消融研究表明:仅含OGT模块的模型(PRIME/-MLM)在ProteinGym与ΔTm基准的零样本表现均不佳;故不采用OGT模块筛选稳定性单点突变位点;建议采用PRIME预测OGT(作为附加预训练任务)时所得的大语言模型(LLM)概率值;
基于生物学家既往研究经验,可选择位于蛋白质表面的突变提升稳定性,并改造蛋白口袋周围氨基酸以增强催化活性。
LbCas12a蛋白
多功能蛋白工程因特性间此消彼长的权衡关系而颇具挑战性;其功能态与晶体结构间显著的构象差异,对依赖结构的传统理性设计方法提出严峻挑战。本研究选用需多功能域协同作用的大蛋白Lachnospiraceae bacterium Cas12a(LbCas12a)验证模型能力。
对LbCas12a开展熔解温度(Tm)工程化改造。Cas12a属V-A型CRISPR-Cas系统的RNA引导内切核酸酶。该蛋白含1228个氨基酸及多个功能域(图3A所示)。催化过程中,CRISPR RNA(crRNA)引导Cas12a结合并切割双链DNA底物;靶DNA识别后,Cas12a识别结构域发生构象变化,激活其切割非特异性单链DNA的反式活性。此特性使LbCas12a在体外诊断应用中极具价值。通过PRIME预测单点突变并实验验证30个位点,其中9个突变体(V936F、I976L、S962K、M957L、M456I、L59K、Y549K、G49K及C1090D)Tm值不低于野生型。
T7 RNA聚合酶
T7 RNA聚合酶源于T7噬菌体,是由883个氨基酸组成的单体酶。自20世纪80年代初应用于RNA合成以来,它已成为分子生物学与遗传工程领域的重要工具。当前广泛应用于体外转录实验、mRNA疫苗生产、等温扩增检测技术等领域。但其应用存在局限性:转录过程会产生双链RNA等免疫刺激性副产物,导致mRNA疫苗生产中需复杂纯化工艺;近期研究表明:将反应温度提升至48°C以上可有效减少副产物。
然而野生型T7 RNA聚合酶约45°C时即解折叠,导致酶活性下降且无法在高温下转录目标产物。因此提升其热稳定性至关重要。
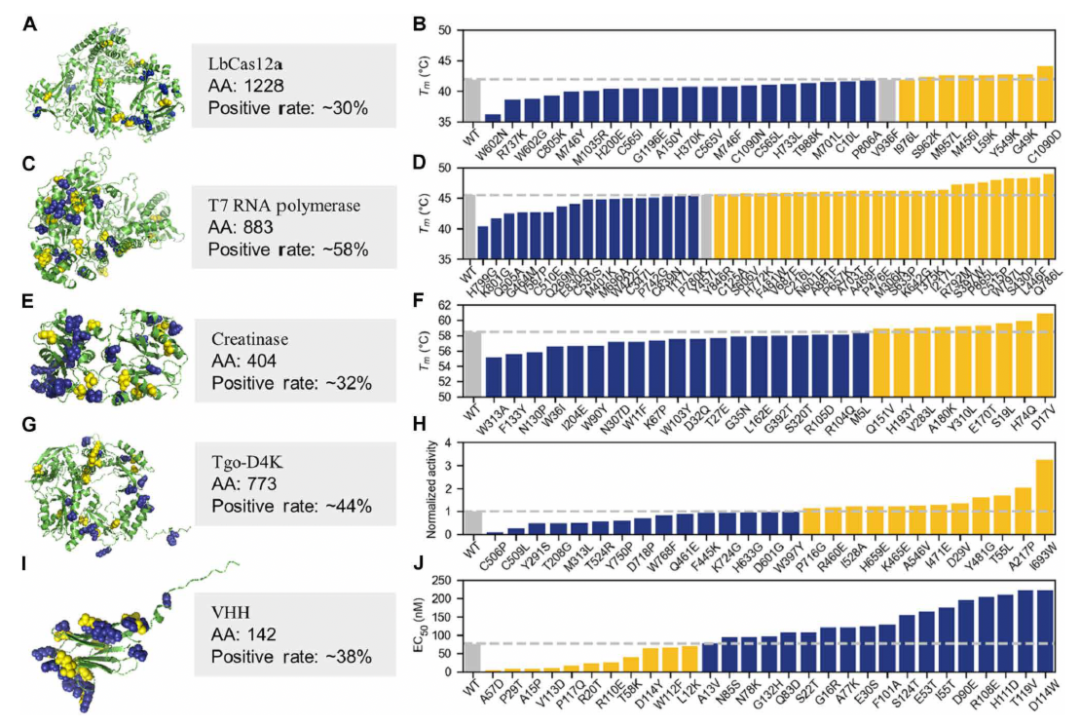
图3. PRIME模型预测单点突变体的结构与性能结果概览。图示PRIME为LbCas12a(A与B)、T7RNA聚合酶(C与D)、肌酸酶(E与F)、非天然核酸聚合酶(G与H)及VHH(I与J)预测的单点突变体结构及实验结果。突变数据点按升序排列,灰色柱标示野生型蛋白基准值以供对比。目标属性优于野生型的突变体标为黄色,劣化突变体标为蓝色。工程目标因蛋白而异:LbCas12a、T7RNA聚合酶及肌酸酶旨在提升热稳定性(Tm);非天然核酸聚合酶(Tgo-D4K)旨在加速FANA合成速率;VHH则需增强极端碱性pH环境耐受性抗原结合中位有效浓度(EC50)。所有突变结构均由AlphaFold2折叠生成。详细实验数据参见补充材料中的独立Excel文件。
本研究通过PRIME预测T7 RNA聚合酶突变位点,并直接筛选出前45个单点突变体进行实验验证。如图3D所示:57.7%(45个突变体中26个)的突变体熔解温度高于野生型(具体突变体:Y846R、C125A、H772K、S606V、V687E、F481W、C216L、N601E、A881F、P657K、S633P、K642G、M306K、A703T、P476E、A468F、T375K、I217L、R792M、S397W、P865L、C515P、W797L、S430P、L446F及Q786L)。
肌酸酶
该二聚体蛋白酶广泛应用于酶法检测肌酐水平;主要源自假单胞菌、芽孢杆菌及产碱菌等微生物;在医学诊断中具有关键作用------用于定量测定血清与尿液中的肌酐含量;肌酐水平升高提示肾脏或肌肉功能受损。然而其最适催化温度仅30-40°C,限制了工业与临床诊断应用;提升热稳定性既可提高临床肌酐检测效率,又利于酶的生产、储存与运输。
本研究采用PRIME模型对源自粪产碱菌的肌酸酶单点突变位点进行预测;最终筛选28个单点突变体开展实验验证;如图3F所示:32%(28个突变体中9个)的热稳定性得到提升(Q151V、H193Y、V283L、A180K、Y310L、E170T、S19L、H74Q及D17V)。
这里面有个问题,预测出的热稳定性排序,与实际实验的热稳定性排序吻合度怎样?
非天然核酸聚合酶
Tgo DNA聚合酶源自新西兰热泉口分离的嗜热古菌Thermococcus gorgonarius。 该酶能精确复制脱氧核糖核苷酸中含2'-氟阿拉伯糖残基的遗传聚合物FANA。但其催化FANA在DNA模板上合成的速率仅~15 nt/min,远低于自身催化DNA合成的速率(~400 nt/min),制约了FANA在信息存储、疾病治疗等领域的DNA替代应用。异源核酸(XNA)聚合酶的进化需全面评估结合亲和力、催化活性及持续合成能力。因XNA具有区别于DNA/RNA的独特化学与生物物理特性,传统计算方法难以预测。XNA特殊的糖环折叠构象可能形成异于DNA/RNA及碱基修饰核酸的空间结构,影响聚合酶对XNA的识别。这使得XNA聚合酶的计算机预测与定向进化仍具挑战性。目前XNA聚合酶进化仅依赖区室化自标记等体外随机突变方法。Pinheiro等通过构建高通量突变库经至少两轮筛选,获得当前最快的FANA聚合酶Tgo-D4K(含突变L403P、P657T等9个位点)。该酶催化FANA在DNA模板上延伸速率达~80±27 nt/min,而DNA延伸速率降至16±3 nt/min(62)。但Tgo-D4K合成FANA的速率仍低于Tgo合成DNA的速率。因此亟需改造现有聚合酶以筛选具更高FANA合成速率的酶变体。
本研究以Tgo-D4K为起点展开探索------通过PRIME系统筛选Tgo-D4K各结构域的潜在突变位点;最终选取27个有前景的突变位点进行实验验证。采用聚合酶动力学分析(PKPro)策略检测突变体FANA合成速率;实验结果(图3H)显示:40%以上突变体(27个中12个)FANA合成速率提升(包括P716G、R460E、I528A、H659E、K465E、A546V、I471E、D29V、Y481G、T55L、A217P及I693W),其中单点突变I693W使延伸速率提升至Tgo-D4K酶的~3.2倍。
VHH抗体
VHH抗体是重链抗体的抗原结合片段;因其尺寸小、单体状态、结构稳定及易于改造等优势,VHH已成为医学研究与临床抗体药物开发的重要工具------作为亲和配体选择性纯化凝血酶原、四溴双酚A、细胞间黏附分子1等生物制剂;生物制品实际生产中,最广泛采用的在位清洗方法为0.5 M NaOH溶液处理24小时;因此需通过突变工程改造VHH抗体,使其耐受自然界罕见的强碱性环境。
本研究通过PRIME模型预测针对生长激素的VHH抗体突变位点(该抗体选自免疫羊驼);筛选排名前29的突变体进行测试:如图3J所示:29个突变体中11个(约38%)在0.3 M NaOH处理24小时后稳定性提升(A57D、P29T、A15P、V113D、P117Q、R20T、R110E、T58K、D114Y、W112F及L12K);其中A57D突变体的耐碱性提升达12倍;此外,约31%(29个突变体中9个)在碱处理前即显示抗原亲和力增强(P29T、A15P、A57D、P117Q、Q83D、R20T、T119V、L12K及V113D)。
单点突变筛选策略的基准测试
为评估PRIME模型及单点突变筛选策略效能,我们建立不同单点突变选择策略的基准对比体系;涵盖计算机模拟与湿实验双重验证。基于ProteinGym数据集筛选单点饱和突变子集(含5个野生型序列与PRIME预训练数据集同源性<30%的数据集)进行分析;对比四种策略筛选的Top15单点突变:
(i) 本论文策略(目标蛋白同源序列微调PRIME);
(ii) 同源序列微调ESM-2;
(iii) 文献(74)的ESM投票策略;
(iv) 随机单点突变。
在阳性突变数量、最大适应度及突变体中位适应度三项指标上,本策略持续领先(详见表S5);通过湿实验对比不同方法所选Top15单点突变性能:聚焦T7 RNA聚合酶与非天然核酸聚合酶Tgo-D4K两种蛋白;验证本策略、ESM投票策略、同源序列微调ESM-2策略及Rosetta解折叠自由能单点饱和突变评分策略的Top15突变体;Rosetta依据解折叠自由能预测值对单点饱和突变排序评分;所用能量函数涵盖文献(75)全部能量项。图4(A与B)及表S6显示:本策略所选单点突变在性能上全面优于其他策略。
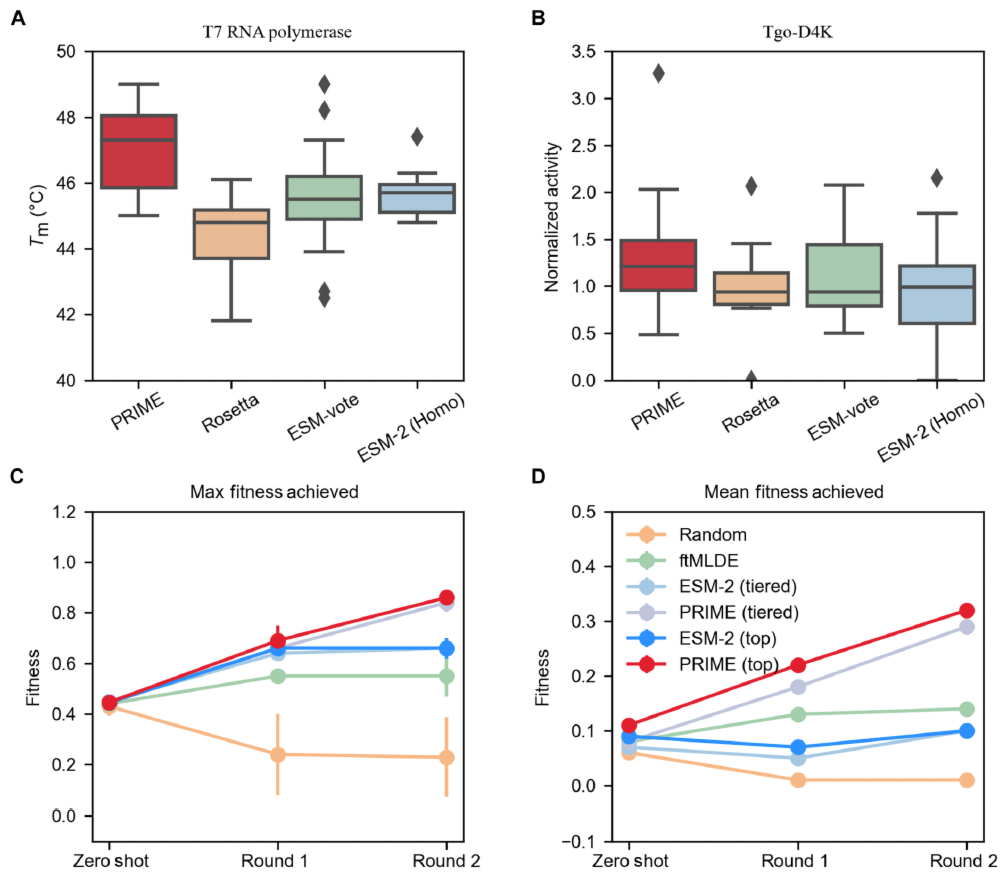
图4. PRIME与其他模型的湿实验与计算机模拟基准对比。(A)T7 RNA聚合酶Top15单点突变的湿实验结果对比;(B)PRIME、Rosetta、ESM投票策略及同源微调ESM-2对Tgo-D4K的评估结果。(C与D)基于GB1数据集的计算机模拟定向进化结果:随机突变、ftMLDE、ESM-2及PRIME在最大适应度(C)和平均适应度(D)上的表现,ESM-2与PRIME同时测试了Top-K采样和ftMLDE(77)的分层采样策略。
基于PRIME驱动蛋白质工程的增强型多位点突变技术
传统蛋白质工程与定向进化技术常采用渐进式策略(类似贪婪算法),通过累积单点突变构建多位点变体;该策略虽广泛应用,却易陷入局部最优陷阱。最优多位点突变体往往并非最佳单点突变体的简单叠加。基于PRIME能力,我们提出先进的多位点突变叠加策略:这种纯数据驱动方法评估2^N种潜在突变体(N为可组合单点突变数量)的全景图,规避传统定向进化因渐进突变陷入局部最优的缺陷。如图1D所示,该策略简化高性能多位点突变预测,大幅减少实验迭代次数。方法核心是通过目标蛋白同源序列微调PRIME的零样本预测流程;既往研究证实同源序列微调的语言模型(PLMs)在低数据量场景表现更优。
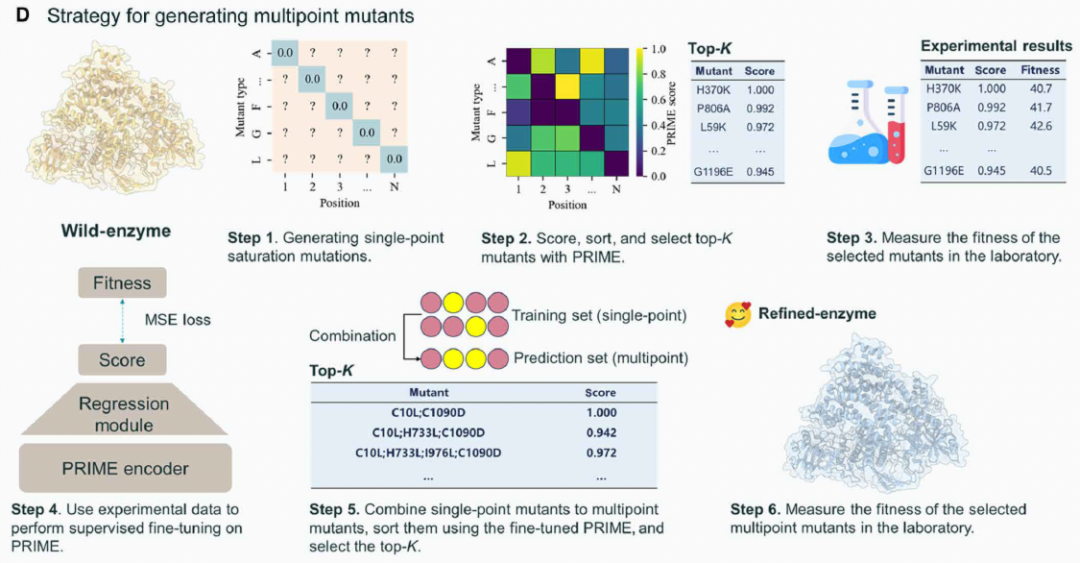
图1. PRIME架构及应用概览。(D)多位点突变体生成策略。第一步:评估单点突变效应,筛选Top-K突变体进行实验适应性检测;第二步:实验数据用于PRIME模型微调,微调后模型预测多位点突变体适应性;第三步:根据预测结果筛选Top-K多位点突变体开展进阶实验。
基于GB1数据集的模拟定向进化对比实验显示:相较ftMLDE方法,PRIME驱动的工作流能更有效筛选最大适应度(图4C)与平均适应度(图4D)提升的多位点突变体。测试比较了PRIME的Top-K采样与ftMLDE的分层采样;研究表明:PRIME迭代多位点突变策略在最大/平均适应度上全面超越ftMLDE;其中Top-K采样效果与分层采样相当且略优(详见表S7)。该计算机模拟定向进化实验执行100次,每次迭代含两轮循环;每轮以前轮筛选的Top50突变体或分层50样本作为训练集,通过多层感知机(MLP)回归模型驱动ESM-2与PRIME评估剩余突变体。 ftMLDE实施采用文献(77)代码,以MSA-transformer为变体编码模型,回归模块集成ARD回归、装袋回归器及K近邻回归器。
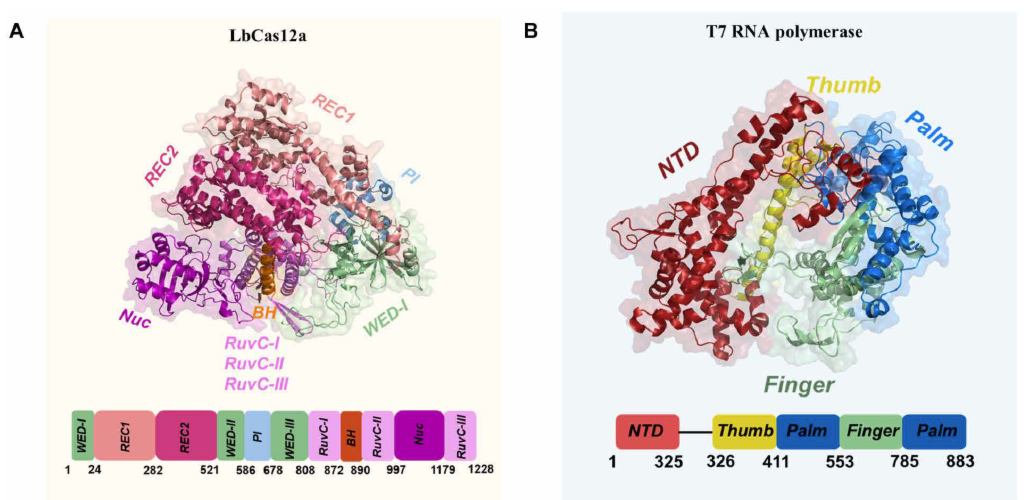
图5. 多位点突变体蛋白结构及实验结果展示(与野生型蛋白特性对比);LbCas12a(AlphaFold2预测结构)(A,C)与T7 RNA聚合酶(PDB ID: 1MSW)(B,D)的多位点突变体蛋白结构及实验结果。 各蛋白结构图下方标示其功能域分布;(C)(D)中红色虚线标示野生型标准化活性水平;五个最优热稳定突变体活性值以红色标记点突出显示,便于与野生型直接对比;野生型热稳定性(Tm)分别以黄色虚线(LbCas12a)和蓝色虚线(T7 RNA聚合酶)标示;R1/R2分别代表第一/二轮多位点组合筛选,后续数字为突变体编号索引。
LbCas12a蛋白研究
基于全部30个单点突变数据点训练PRIME模型,预测多位点突变组合的Tm值;随后从二至四位点突变库中各筛选评分前10的突变体,开展第二轮实验验证;第三轮稳定性进化中,将30个多位点突变体加入训练集进一步微调PRIME;最终从三至六位点突变库中各优选前5个突变体,七至十位点突变库中筛选共10个顶级突变体进行实验表征。
如图5C所示:第二轮测试中30个多位点突变体中的17个Tm值高于野生型;第三轮实验中所有30个多位点突变体Tm值均超越野生型;最优突变体为八位点突变体R2-26------Tm值达48.15°C,较野生型提高6.25°C(详见补充材料)。
在第二轮提升Tm的多位点阳性突变筛选中,PRIME推荐的多数多位点突变体包含Tm降低的负向单点突变。 例如R1-3突变体(C10L; S962K)中的C10L位点Tm低于野生型,但由其构成的双位点突变体Tm却高于两个单点突变体。进一步地,在R1-9双点突变(S962K; I976L)基础上添加C10L形成的三位点突变体R1-15(C10L; S962K; I976L),其Tm值仍高于前代双点突变体。 值得注意的是,含C10L的阳性多位点突变由不同功能域突变组成:如图5A所示,C10L位于Cas12a的WED-I结构域,而S962与I976则位于RuvC-II结构域。这彰显了PRIME卓越的泛化能力------仅凭序列信息即掌握跨结构域突变间的上位效应,并能将负向单点突变组合成优质多位点突变体。传统定向进化采用贪婪算法式渐进策略(仅累积阳性单点突变构建多位点变体),难以实现此类突破。因其几乎不可能直接组合负向单点突变形成多位点突变体。
T7 RNA聚合酶
以T7 RNA聚合酶为例:在已有单点突变数据基础上,采用经T7 RNA聚合酶同源序列微调的PRIME模型执行监督回归预测任务(方法详见原文)。
我们使用第一轮所有单点突变(含野生型)的熔解温度数据作为训练集,利用训练后的PRIME模型预测多位点突变序列;随后从含2-4个突变位点的序列中各筛选预测Tm最高的5个序列,针对8个突变位点选择预测Tm最高的10个序列,共获得25个多位点突变体用于第二轮湿实验验证;如图5D所示:经两轮突变筛选,全部25个多位点突变体熔解温度均高于野生型;最优突变体为含8个突变位点的R1-21(Q786L;S430P;W797L;P657K;N601E;L446F;P476E;T375K),其熔解温度较野生型提升7.4°C。
相较于New England Biolabs商用热稳定型T7RNA聚合酶(Hi-T7,Tm=56.8°C),我们开发的八位点突变体Tm值仍低4°C。
为获得更高Tm突变体,我们追加测试了PRIME首轮零样本预测的10个单点突变体(图5D)。整合全部单点突变体及既往多位点突变体数据(共80个突变体)训练PRIME模型;随后基于训练模型直接预测由这些单点突变形成的9-14位点突变体Tm值;筛选PRIME评分最高的15个多位点突变体进行湿实验验证;其中5个深度突变体Tm明确超越Hi-T7------其中最佳突变体(含12个突变位点:Q786L、S430P等)Tm值较野生型提高12.8°C;值得注意的是:如图5D所示,这五个最优热稳定突变体的酶活性同样高于野生型。
该12位点突变体包含A124N、G618E、L665D等多个负向单点突变;如图5D所示:这些突变若单独作用于野生型将导致熔解温度降低;负突变的组合极具挑战------由于缺乏有效利用的领域知识,传统蛋白质工程常预先排除这些突变位点以构建深度突变体;鉴于负突变发生率远高于正突变,本研究发现蛋白质大语言模型能将负突变作为组分构建更优深度突变体,这对蛋白质工程界深入机制研究与工业应用具有重要启示意义。
在无预实验数据或高通量筛选技术条件下,经过三轮突变筛选及95个突变体的湿实验验证,我们成功获得含12个突变位点的T7 RNA聚合酶变体,其性能超越商业酶;该成果不仅验证了PRIME单点预测与多位点叠加的精准高效,更彰显其显著降低湿实验成本的潜力;这一成就是传统蛋白质工程与理性设计难以企及的。
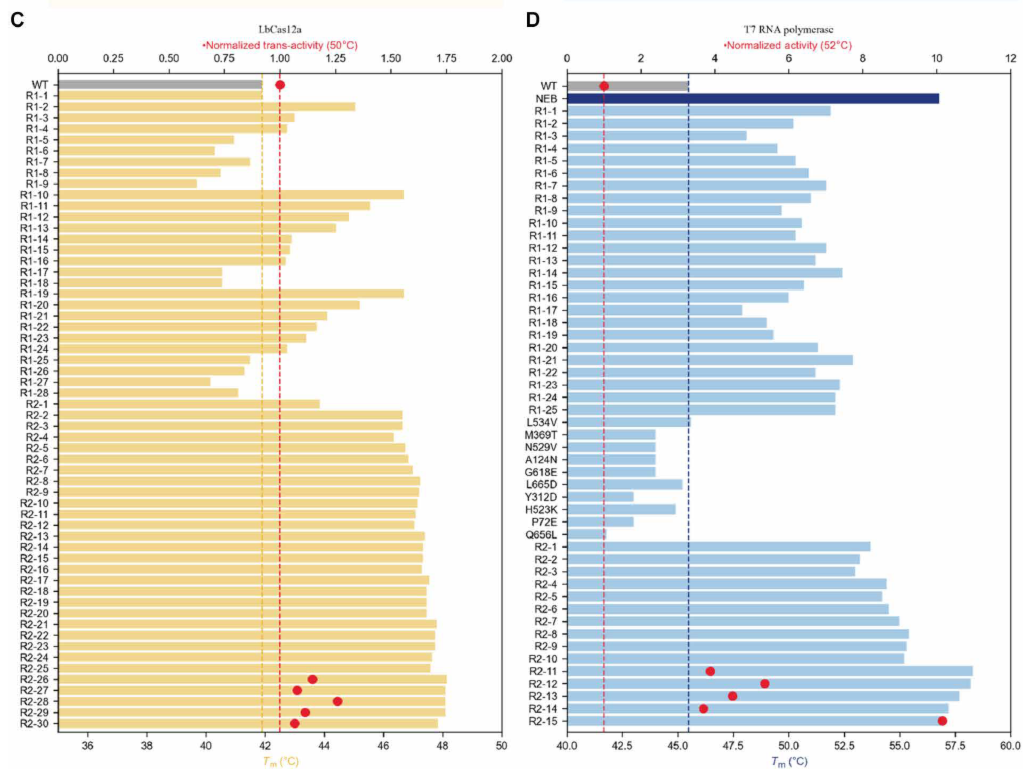
图5. 多位点突变体蛋白结构及实验结果展示(与野生型蛋白特性对比);(C)(D)中红色虚线标示野生型标准化活性水平;五个最优热稳定突变体活性值以红色标记点突出显示,便于与野生型直接对比;野生型热稳定性(Tm)分别以黄色虚线(LbCas12a)和蓝色虚线(T7 RNA聚合酶)标示;R1/R2分别代表第一/二轮多位点组合筛选,后续数字为突变体编号索引。
讨论
我们提出PRIME------一种先进深度学习技术,能巧妙利用涵盖序列-宿主菌株最佳生长温度的庞大数据集。通过为最佳生长温度预测定制掩码语言模型,PRIME精准捕捉了蛋白质序列的语义、语法及温度相关特征。严格的计算机评估一致证明,在预测蛋白质突变体的热稳定性和整体适应性方面,PRIME显著优于ESM-1v、ESM-2、MSA-transformer、Tranception-EVE、CARP、MIF-ST、SaProt、Stability Oracle、GEMME和Rosetta等主流模型。借助PRIME,我们设计了五个单点突变蛋白质,其理化特性显著增强;在30至45个AI构思的突变体中,成功率超30%。这彰显了PRIME在蛋白质工程领域的变革性潜力。
传统上,蛋白质工程策略主要围绕定向进化或理性设计展开。前者虽有效,但依赖高通量实验筛选,在时间和资金上均耗费巨大。对于许多关键蛋白质,高通量实验方法的实用性存疑,使得低通量检测成为更可行的替代方案。反之,理性设计要求深入理解与目标蛋白作用机制相关的生物物理特性。在透彻掌握该机制时,理性设计或可通过有限湿实验鉴定出高性能突变体。然而,对于作用机制认知有限的蛋白质或非常规活性改造(如本研究强调的针对非天然核酸的聚合活性),理性设计常遇瓶颈。在此类情境中,以PRIME为代表的人工智能预测方法脱颖而出。无需大量湿实验数据或对蛋白作用机制的深刻理解,PRIME即可提供宝贵洞见,优化蛋白质工程路径。
传统蛋白质工程常采用类似贪婪算法的策略,从单点突变逐步积累成多位点突变体。尽管有效,但该过程通常耗时费力,且偶现次优结果------最优多位点突变体未必由最有利的单点突变组成。我们的PRIME模型为此领域引入范式转变:提供优化的多位点突变积累策略,突破传统方法局限并加速优质多位点蛋白突变体的创制。PRIME能将负面单点突变自动整合为深度突变体,显著提升其适应性。这一发现意义重大,为蛋白质工程师开辟新路径------他们可利用比正向突变更普遍、曾被传统设计排除的负面突变增强蛋白适应性。通过降低对大量实验筛选的依赖,PRIME等计算工具有望革新蛋白质工程领域,拓展可精密改造的蛋白范围,为制药与工业应用带来广阔前景。
此外,PRIME的多功能建模框架有望支持多样化的预测任务,例如推断天然蛋白质的解链温度(Tm)或酶最适活性温度(Topt)。PRIME简化了蛋白质修饰的先决条件,促进蛋白质稳定性与活性的增强,省却全面作用机制探查的需求。PRIME的多任务学习模式将最佳生长温度预测与掩码语言模型对齐,相较其他训练技术,显著提升了模型在温度相关下游任务的预测精度,且不影响其非温度关联任务的预测效力。这表明:该预训练方法在增强模型特定任务预测能力的同时,也维持了其在其他无关任务上的泛化能力。这种预训练方法或可开辟全新学习范式,将专业领域洞见融入基础AI框架,助力弥合深度学习与传统科学认知的鸿沟。PRIME的预测能力还延伸至精准定位增强蛋白质特性的突变位点(包括自然界罕见的位点),例如强化抗体在极端碱性环境的耐受性,或提升聚合酶对非天然核酸的聚合速率,彰显了PRIME在蛋白质工程中的普适性。
方法
PRIME架构细节
PRIME包含一个通用Transformer编码器和两个独立组件:其一执行掩码语言模型预训练,其二用于最佳生长温度预测预训练。本节首先阐述通用Transformer编码器,随后详解掩码语言模型模块与最佳生长温度预测模块。
Transformer编码器
本模型采用广泛使用的预训练语言模型ESM-2的Transformer架构。相比标准Transformer架构,其将绝对位置嵌入替换为旋转位置嵌入,采用Roberta式的预层归一化技巧 (prelayer normalization),激活函数使用GELU而非ReLU。我们还应用Flash注意力机制加速训练与推理,代码详见代码库。该Transformer编码器本质是参数化转换函数,可将蛋白质序列转化为稠密向量序列 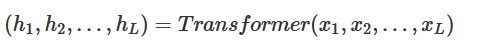

MLM模块
本模块架构同ESM-2,充当Transformer编码器的逆向函数,将隐向量序列映射为蛋白质序列的独热编码。预训练中,MLM模块学习恢复噪声化蛋白质序列:该噪声序列通过随机掩蔽蛋白质序列中20%的token生成,其中70%被掩蔽token(占序列总长度的14%)替换为特殊
OGT预测模块

随后采用包含两个全连接层和GELU激活函数的MLP层转换加权向量c:第一全连接层将c映射至Transformer前馈网络层同等维度(本实现中为隐藏层大小的四倍),第二全连接层将首层输出映射回原维度。两层间设GELU激活函数,并在第二全连接层输出与注意力层输出间构建残差连接 

单点突变效应的零样本预测
基于文献,经MLM目标训练的蛋白质语言模型能预测特定蛋白位点氨基酸出现的概率。此预测能力可用于评估序列突变效应:图1C展示MLM模块预测突变效应的方法。给定突变中,野生型蛋白的氨基酸作为参照状态,通过比较突变氨基酸与原始(野生型)氨基酸的预测概率确定突变效应。量化公式为突变位点的对数比值比: 
训练细节
预训练流程
如图1A所示,PRIME模型预训练期间融合三项损失函数作为优化目标:掩码语言建模(MLM)损失、最适生长温度(OGT)预测损失及相关性损失。各函数数学定义如下(预训练中损失变化曲线见图S1)。
掩码语言建模损失:采用交叉熵损失函数。对蛋白质序列中每个被屏蔽氨基酸,模型计算其在20种天然氨基酸词汇表上的概率分布,并与真实氨基酸的独热向量分布进行比对:
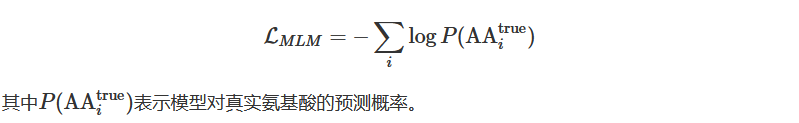
OGT预测损失
该损失函数量化预测的最适生长温度(OGT)与实际OGT的偏差,采用均方误差(MSE)作为损失函数:
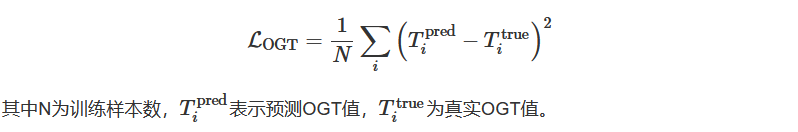
相关性损失

最终损失为三项模型损失之和。我们观察到OGT预测损失的量级显著高于其他两项损失,其值初始范围0-1000,后期稳定在0-100。为确保数值稳定性,我们对该损失乘以0.01进行加权。
实现细节
我们使用PyTorch实现PRIME。Transformer编码器含33层和20个注意力头,参数量6.5亿,嵌入尺寸1280。学习率设为 1 * 10^{-4},单GPU微批次大小为4096 tokens,梯度累积步数为32。模型在8×A100 80G GPU上训练20万步更新。预训练后,OGT预测任务在5万保留验证集的均方根误差为3.5,MLM困惑度达3.52,关联损失平均误差为0.1623。Transformer编码器及MLM模块所有层初始化参照文献(19)。
交替训练
因三大损失函数输入要求差异(MLM损失处理噪声化蛋白质序列,OGT损失需完整序列,关联损失需序列的N个随机单点突变体),我们采用交替训练策略优化不同目标。具体使用Adam优化器的Mini-batch梯度下降法训练模型,每轮mini-batch迭代切换任务。训练方案详见附表S8的Python与PyTorch伪代码。训练后预测OGT与实际OGT对比结果见图S2。
多任务损失函数权重对零样本预测性能的影响
我们通过调整损失函数权重探索多任务损失函数设计,以平衡数据量与任务难度差异。为降低计算成本,从9600万条预训练数据中随机选取50万条序列作为消融研究的训练集。通过网格搜索从权重列表0.01, 0.05, 0.5, 1, 2中选择组合,共生成125组权重配比。发现1:1:1权重比在ProteinGym及ΔTm数据集上为零样本突变预测任务的最优配置,具体结果记录于附表S9。
同源序列微调MLM模块
为提升PRIME与ESM-2的零样本突变效应预测性能,我们探索仅基于MLM的同源序列微调优化策略。该微调通过利用相似蛋白序列知识,使预训练模型适配特定蛋白质。此策略应用于ProteinGym或Tm数据集:采用知名序列比对工具Jackhammer从Uniclust30数据库识别数据集内蛋白的同源序列。同源序列超3万条的蛋白仅取前3万条,不足3万条则保留全部用于微调。微调过程复用MLM预训练阶段的超参数设置:噪声化序列通过随机掩蔽蛋白序列20%的token生成,其中70%掩蔽token(占全序列14%)替换为
PRIME在温度相关基准与FLIP数据集上的迁移学习
PRIME同时训练于温度相关的监督与非监督任务。为评估其迁移表征能力,采用解链温度(Tm)预测基准与最适催化温度(Topt)预测基准。评估使用编码器组件与OGT模块,Transformer编码器所有参数均可微调:批次大小为256,Adam优化器学习率0.0001;模型采用早停策略(耐心值20轮),最大训练轮数200轮,损失函数同为MSE。为确保鲁棒性,实验采用五折交叉验证,且保证测试集未包含训练验证阶段信息。最终性能指标取结果均值,方差用于生成误差棒。
PRIME在监督式突变效应预测中的迁移学习
PRIME同样适用于监督式突变效应预测任务(用于生成多位点突变体策略,见图1D)。给定含实验适应性标签的突变序列训练集,PRIME通过训练集学习并预测新突变序列的适应性。本任务仅使用Transformer编码器与OGT模块(弃用MLM模块)。除OGT模块末两层全连接层(FC_3与FC_4)参数重新初始化外,其余参数冻结(即图1D回归模块)。采用MSE损失最小化预测与真实适应性的差异:学习率 `1 x 10^{-4} \,批次大小16。
训练轮数动态确定:先将数据集五等分,每次迭代以4份训练、1份验证;记录五次验证所需轮数,取其平均值作为最终训练轮数。最终训练阶段不使用验证集,而是以该轮数在全数据集上训练。训练后模型可预测未见过突变体的适应性。对于无标记突变数据的蛋白质,首先利用PRIME的零样本能力(PRIME Zero-shot)筛选top-K单点突变体进行实验标记。第一轮设计中,该标记数据用于训练PRIME(称为PRIME第一轮),训练后的模型预测零筛轮所得单点突变体组合成的多位点突变体适应性,据此得分选择top-K突变体实验测定其适应性。将首轮标记的多位点突变体加入初始训练集,重训PRIME(即PRIME第二轮)。利用PRIME第二轮预测零筛轮与第一轮突变体组合的所有多位点突变体适应性,筛选新top-K突变体实验测定。若结果未达要求,则将标记的top突变体加入训练集重复流程。
数据集
预训练数据集
通过整合Uniprot公开数据及宏基因组项目蛋白序列,我们构建了ProteomeAtlas------含47亿天然蛋白序列的大型数据库。筛选时仅保留完整长度序列,采用MMseqs2工具以50%序列同一性阈值去除冗余,鉴定并注释了菌株最佳生长温度。最终完成9600万条序列注释,为探索蛋白质序列-温度关系提供丰富资源库。
零样本突变评分基准数据集
解链温度变化(ΔTm)数据集源自MPTherm、FireProtDB及ProThermDB,确保所有实验在相同pH条件下进行。ProteinGym数据集引自(31)。预测天然蛋白质序列解链温度(Tm)与最适酶活温度(Topt)的数据集及划分摘自(82)。
面向不同工程目标的单点突变筛选策略
PRIME可基于活性和稳定性对单突变体排序。但PRIME消融研究表明,仅含OGT模块的模式(PRIME/-MLM)在ProteinGym基准和ΔTm任务中零样本性能均较差,故不推荐使用OGT模块筛选稳定性相关的单点突变。建议采用PRIME在OGT预测任务中获得的语言模型似然值。根据生物学研究经验:选择蛋白表面的突变位点可提升稳定性且对活性影响甚微;突变蛋白口袋周围的氨基酸位点则可增强催化活性。此类经验法则应用于特定蛋白质工程任务,或可进一步提升成功率。
五类蛋白质高稳定性与高活性的工程化改造
PRIME单点突变预测流程
首先,使用Jackhmmer从Uniclust30数据库中识别各靶蛋白的同源序列。同源序列超3万条的蛋白质,随机筛选3万条用于PRIME模型微调;不足3万条则全部纳入微调。每个靶蛋白进行五轮微调(每轮以不同随机种子初始化)。整合五组参数的单点饱和突变预测结果,生成各蛋白的突变体评分表。评分超过野生型的突变体标记为候选对象,最终人工精选约30至45个突变体------确保其位于催化活性位点或结合口袋等关键区域的6埃半径外,以供后续实验验证。
T7 RNA聚合酶
突变体制备流程:将T7 RNA聚合酶(Uniprot ID: P00573)基因及其突变体基因克隆至pQE-80l表达载体,转化大肠杆菌BL21(DE3)。细胞在LB培养基中培养至600 nm波长光密度(OD600)达0.6--0.8,加入1 mM异丙基-β-d-硫代半乳糖苷(IPTG)于37°C诱导表达6小时。收集菌体后重悬于结合缓冲液50 mM tris-HCl (pH 8.0), 300 mM NaCl, 3 mM咪唑, 0.1 mM EDTA,超声破碎裂解;裂解液4°C、12,000rpm离心30分钟;上样至镍-氮川三乙酸(Ni-NTA)重力柱,用洗涤缓冲液50 mM tris-HCl (pH 8.0), 300 mM NaCl, 10 mM咪唑, 0.1 mM EDTA, 10%甘油冲洗;以洗脱缓冲液50 mM tris-HCl (pH 8.0), 300 mM NaCl, 250 mM咪唑, 0.1 mM EDTA, 10%甘油洗脱目标蛋白;最后用超滤缓冲液50 mM tris-HCl (pH 8.0), 100 mM NaCl, 0.1 mM EDTA浓缩,并以储存缓冲液50 mM tris-HCl (pH 8.0), 100 mM NaCl, 0.1 mM EDTA, 1 mM二硫苏糖醇(DTT), 75%甘油稀释(83)。
热熔解实验检测:蛋白染色剂SYPRO橙染料以5×终浓度添加,蛋白样品(浓度约0.2 mg/ml)混合于八联排PCR管中。每样本终体积20 μl,设三组重复。使用PCR仪(Analytik Jena qTower3)生成变性曲线,该设备配备激发波长470nm(FAM)/发射波长625nm(ROX)光学滤光片。温度从25°C以0.5°C步长升至65°C;每步平衡5秒。通过玻尔兹曼方程拟合热解折叠曲线计算Tm近似值(58)。
体外转录(IVT)反应检测:配制含200 mM Hepes (pH 7.5)、30 mM MgCl₂、20 mM DTT、核糖核酸酶抑制剂(0.4 U/μl)、5 mM核苷三磷酸混合物及100 nM iSpinach DNA模板(84)的IVT反应缓冲液。该缓冲液52°C孵育10分钟,加入0.04 mg/ml T7 RNA聚合酶启动反应;反应液孵育1小时后添加100 mM EDTA终止反应;最终加入100 μM DFHBI荧光染料,于470 nm激发光/512 nm发射光波长下检测荧光强度。
肌酸酶
突变体制备流程:将肌酸酶(Uniprot ID: Q9RH‑U9)基因克隆至pET-28a表达载体,转化大肠杆菌BL21(DE3)。表达肌酸酶的细胞在LB培养基中于37°C、220rpm震荡培养;当培养液OD600值达0.8--1.0时,添加1 mM IPTG诱导表达,随后在18°C继续培养16小时。收集细胞后重悬于结合缓冲液25 mM tris-HCl (pH 8.0), 200 mM NaCl, 20 mM咪唑,超声破碎;裂解液经4°C、12,000 rpm离心30分钟取上清;上清液上样至预平衡的镍-氮川三乙酸重力柱,采用20至200 mM的咪唑梯度洗脱目标蛋白;最终通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分析洗脱组分的纯度。
合并含目标蛋白的纯化组分,通过超滤装置脱盐;浓缩纯化蛋白后,储存于1× PBS缓冲液中(-80°C条件下保持稳定性与活性)(58)。
差示扫描荧光法:热稳定性检测同样使用PCR仪(Analytik Jena qTower3)。所有蛋白于1 × PBS缓冲液稀释至终浓度0.3 mg/ml,在八联排PCR管中与5×终浓度的SYPRO橙染料混合。对样品进行25至85°C范围的热处理(每步升温0.5°C,每步平衡5秒)启动蛋白解折叠过程;获取热解折叠曲线后,采用玻尔兹曼方程分析数据计算Tm值(58)。
酶活检测:肌酸酶将肌酸水解产生尿素和肌酐;尿素与对二甲基氨基苯甲醛反应生成黄色染料,通过分光光度计测定435 nm波长下该染料的吸光度可计算尿素浓度(58),进而推算蛋白比活性。实验步骤如下:
-
将含100 mM肌酸的PBS缓冲液(280 μl)于37°C孵育5分钟;
-
加入20 μl蛋白溶液(1 mg/ml)继续孵育22分钟;
-
加入终止反应液------2 g对二甲基氨基苯甲醛溶于100 ml二甲基亚砜与15 ml浓盐酸配制的溶液(600 μl);
-
使用分光光度计检测435 nm处吸光度。
VHH纳米抗体
蛋白表达与纯化:将VHH基因克隆至带N端His标签的pET29a质粒,转化大肠杆菌BL21(DE3)。重组菌单菌落接种含卡那霉素(50 μg/ml)的30 ml LB培养基,37°C摇床培养12--16小时作种子液;取10 ml种子液转接含50 μg/ml卡那霉素的1 L LB培养基,37°C、220rpm震荡培养至OD600值0.6--0.8;冷却至16°C后加入0.5 mM IPTG,16°C诱导表达20--24小时。发酵液4000 rpm离心30分钟收获细胞沉淀用于纯化:沉淀重悬于缓冲液A20 mM Na₂HPO₄/NaH₂PO₄与0.5M NaCl (pH 8.0),超声裂解;裂解液4°C、12,000rpm离心30分钟,上清经镍-氮川三乙酸亲和层析纯化(洗脱缓冲液:20 mM Na₂HPO₄/NaH₂PO₄、0.5M NaCl、250 mM咪唑 (pH 8.0))。十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分析洗脱组分纯度,合并含目标蛋白的组分经超滤装置脱盐;浓缩蛋白后于缓冲液A(添加10%甘油)中-80°C储存。
蛋白质碱处理:野生型及VHH突变体于0.3或0.5摩尔NaOH溶液中孵育3/6/24小时;加入盐酸终止碱处理后,样本−80°C冻存。
碱性pH稳定性检测(ELISA):96孔板每孔包被5 ng生长激素蛋白,4°C过夜孵育;PBST缓冲液洗涤三次;1× PBST含1% BSA溶液25°C封闭2小时后,以1× PBST洗涤三次;加入系列稀释VHH蛋白(100 μl/孔,稀释梯度:1:2、1:4、1:8、1:16、1:32、1:64、1:128、1:256、1:512、1:1024、1:2048),25°C孵育1小时;0.5% PBST洗涤三次,添加辣根过氧化物酶(100 μl/孔,1:5000稀释)25°C孵育1小时;1× PBST洗涤四次后加入TMB底物(100 μl/孔),25°C避光孵育15分钟;最终加入2摩尔硫酸(H₂SO₄,100 μl/孔)终止反应,450 nm波长下检测吸光度(瑞士TECAN仪器)。
通过分析对数激动剂浓度-响应曲线(四参数变量斜率模型),计算半数有效浓度以确定VHH经碱处理后的稳定性。
非天然核酸聚合酶
表达与纯化流程:聚合酶按文献方法表达纯化(85)。简述如下:Tgo-D4K及其突变体基因克隆至pGDR11载体,转化大肠杆菌BL21;菌液于含100 μg/ml氨苄青霉素的50 ml LB培养基中37°C、240rpm震荡培养至OD600值0.6--1.0;加入0.5 mM IPTG后,16°C、240rpm震荡诱导20小时。离心收集细胞,沉淀经裂解缓冲液10 mM tris-HCl (pH 8.0)、500 mM KCl、10%甘油超声破碎;裂解液4°C、13,300rpm离心30分钟,取上清80°C热处理1小时后立即冰浴冷却30分钟;复经4°C、13,300rpm离心30分钟澄清。添加0.5%(v/v)聚乙烯亚胺沉淀核酸,裂解液再次4°C、13,300rpm离心30分钟;加入60%(w/v)硫酸铵沉淀聚合酶,4°C孵育1小时后同条件离心。蛋白沉淀重悬于预冷缓冲液10 mM tris-HCl (pH 8.0)、50 mM KCl、10%甘油,上清上样镍-氮川三乙酸树脂;200 mM咪唑洗脱的蛋白于4°C缓冲液10 mM tris-HCl (pH 8.0)、50 mM KCl、10%甘油透析;洗脱组分经十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)验证纯度后,−80°C储存。
聚合酶合成速率检测:按文献方法(62)进行动力学测量以测定聚合酶合成速率。每反应体系(10 μl)含:1 μM 30聚体模板、100 μM各核苷三磷酸、1× ThermoPol缓冲液、2× LC Green Plus荧光染料及20 nM聚合酶。反应液95°C变性3分钟,55°C延伸30分钟,每6秒间隔记录荧光强度;各聚合酶荧光数据经归一化转换为每聚合酶核苷酸数量,通过线性拟合反应时间内核苷酸累积量计算合成速率,最终值为三次独立重复实验的平均值。
LbCas12a
质粒构建:通过重叠延伸PCR技术构建LbCas12a突变体,以含有野生型LbCas12a的pET28a质粒为模板,搭载目标突变位点的寡核苷酸为引物;表达质粒含C端10× His标签用于后续亲和纯化。重组质粒转化至大肠杆菌Trelief 5α感受态细胞(中国北京Tsingke公司),所有质粒构建体经桑格测序(Tsingke)验证序列。
蛋白表达与纯化:所有LbCas12a蛋白于含卡那霉素(50 μg/ml)的LB培养基中通过大肠杆菌BL21(DE3)表达。从LB琼脂平板挑取单菌落过夜培养制备种子液;次日按1:100比例接种含50 μg/ml卡那霉素的新鲜LB培养基,37°C培养至OD600值达0.6;添加1 mM IPTG于37°C诱导表达4小时;4°C条件下5000 rcf离心15分钟收集细胞。
收集的细胞重悬于裂解缓冲液(pH 8.0)含100 mM磷酸钠、600 mM NaCl、0.05% Tween 20、30 mM咪唑、1 mM二硫苏糖醇、0.5 mM苯甲基磺酰氟;超声破碎后经4°C、12,000 rcf离心1小时;依据制造商方案采用HisPur镍-氮川三乙酸磁珠(美国赛默飞世尔科技公司)纯化蛋白;纯化蛋白通过Pierce蛋白浓缩仪(赛默飞世尔)浓缩至储存缓冲液50 mM tris-HCl (pH 7.5)、500 mM NaCl、10%(v/v)甘油、2 mM二硫苏糖醇,−80°C储存。
crRNA制备:本研究所有DNA寡核苷酸购自Tsingke公司。crRNA制备中,IVT模板通过退火搭载T7启动子的寡核苷酸链与含反义T7启动子、crRNA直接重复基序及间隔序列的互补链生成;使用上述模板及HiScribe T7快速高产量RNA合成试剂盒(New England Biolabs)进行30微升体系转录反应,37°C过夜孵育;经脱氧核糖核酸酶I(0.08 U/μl)处理移除残留DNA模板,最终通过TRIzol试剂(Invitrogen)纯化RNA产物。
差示扫描荧光法检测:所有LbCas12a蛋白在反应缓冲液50 mM tris-HCl (pH 7.5)、500 mM NaCl中稀释至终浓度0.5 mg/ml,加入标准毛细管(NanoTemper);实验在20°至95°C范围内通过Prometheus NT.48仪器及PR.ThermControl软件(德国慕尼黑NanoTemper)进行,升温速率1°C/分钟。
体外切割实验:Cas12a反式切割反应基本参照文献(86),略有优化。靶DNA通过质粒PCR扩增(特异性引物)或退火寡核苷酸制备后纯化。简要流程:10微升反应体系含50 nM LbCas12a蛋白、2.5 ng底物DNA、100 nM crRNA、0.5 mM二硫苏糖醇、1.25 μM单链DNA(5′-FAM-CCC‑CC-BHQ1-3′)及1×缓冲液2.1(新英格兰生物实验室);每样本设三次生物学重复,加样至384孔板;42°C孵育15分钟后,使用SpectraMax iD3多功能酶标仪(激发波长485 nm/发射波长535 nm)监测荧光强度;荧光信号每2分钟记录一次,用于后续分析。
高颜值免费 SCI 在线绘图(点击图片直达)
最全植物基因组数据库IMP (点击图片直达)

往期精品(点击图片直达文字对应教程)
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|
|  |
|  |
|  |
|  |
|