接上节(scanpy单细胞转录组python教程(一):不同形式数据读取)。scanpy流程和R seurat流程并无太多差别,只是语言变了而已,思维不变。读取完矩阵,那么质控无非就是对于UMI、features、线粒体基因、核糖体基因比例、双细胞等等的检测而已。这里我们演示的数据是之前R里面演示过的数据。这里仅仅分析单个样本,不涉及到数据合并,去批次等等,主要是为了熟悉scanpy流程和代码,实际情况可能你不止一个样本,多样本数据分析我们后续讲到。学会基础,后面的也不再话下!
数据质控之数据分析之基,是非常重要的一个步骤,这个步骤也是比较主观的步骤,需要结合实际,可能很多情况需要在做好分群结果之后返回重新调整。这里介绍基础的质控,还有去除背景/环境RNA、去除双细胞等也是质控,后续我们也会涉及到。
import pandas as pd
import scanpy as sc
sc.settings.verbosity = 3
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor="white")read data:这里演示的数据是一个10X数据,经典三个文件表达矩阵!
adata = sc.read_10x_mtx(
"./单样本数据/AA_scRNA/", # the directory with the `.mtx` file
var_names="gene_symbols", # use gene symbols for the variable names (variables-axis index)
cache=True, # write a cache file for faster subsequent reading
)
adata
... writing an h5ad cache file to speedup reading next time
Out[3]:
AnnData object with n_obs × n_vars = 11967 × 33538
var: 'gene_ids', 'feature_types'基础过滤QC:执行最简单的过滤,每个细胞至少表达200个基因,每个基因至少有3个细胞表达。Seurat中这一步的过滤在creatseuratobj的时候就可以选:WT <- CreateSeuratObject(counts = WT, project = "WT", min.cells = 3, min.features = 200)
#basic QC
sc.pp.filter_cells(adata, min_genes=200)#each cell expres 200 genes at least
sc.pp.filter_genes(adata, min_cells=3) #each gene must expres in 3cells at least
filtered out 1149 cells that have less than 200 genes expressed
filtered out 11516 genes that are detected in less than 3 cells然后就是几个我们比较关心的指标了,线粒体基因表达比例,核糖体基因表达比例,血红蛋白基因表达比例。需要注意物种,人和小鼠,人的一般基因大写,小鼠小写。
adata.var["mt"] = adata.var_names.str.startswith("MT-")#开头是MT-的基因
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
adata.var["hb"] = adata.var_names.str.contains(("^HB[^(P)]"))
#Calculate quality control metrics.
#in this step, QC metrics, including 'n_genes_by_counts','total_counts','pct_counts_mt','pct_counts_ribo','pct_counts_hb' & etc, are calculated.
#The funcion sc.pp.calculate_qc_metrics was employed to calculate.
#Pay attention, the parameters percent_top & log1p are optional.
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt", "ribo", "hb"], inplace=True, percent_top=[20], log1p=True)
adata
#######################################################################
AnnData object with n_obs × n_vars = 10818 × 22022
obs: 'n_genes', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'me计算完成后,adata将包括这些内容:
adata.obs:
total_counts:每个细胞的总 UMI 数
n_genes_by_counts:每个细胞检测到的基因数
pct_counts_mt:线粒体基因表达占比(%)
pct_counts_ribo:核糖体基因表达占比(%)
pct_counts_hb:血红蛋白基因表达占比(%)
percent_top_20:表达量最高的 20% 基因的占比(%)
adata.var:
n_cells_by_counts:表达该基因的细胞数
mean_counts:该基因的平均表达量
pct_dropout_by_counts:未检测到该基因的细胞百分比
#obs里面移除一些没用的列,精简数据
remove = ['total_counts_mt', 'log1p_total_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb']
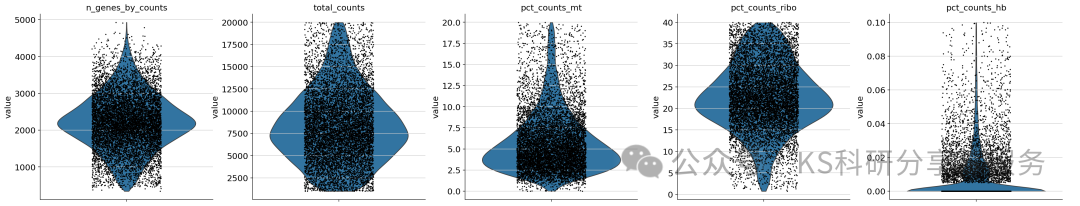
adata.obs = adata.obs[[x for x in adata.obs.columns if x not in remove]]plot quality control metrics,小提琴图可视化,这里是看一下数据分布。
sc.pl.violin(
adata, #adata object
["n_genes_by_counts", "total_counts", "pct_counts_mt",'pct_counts_ribo', 'pct_counts_hb'], #adata.obs或者adata.var_names需要plot的值
jitter=0.2,#抖动点宽度
size=2,#点的大小
multi_panel=True
)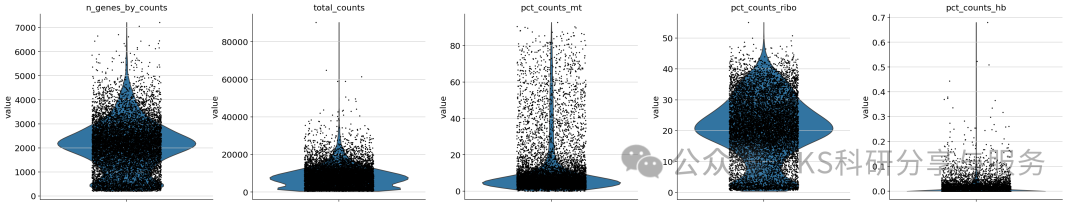
数据QC: 数据的质控要结合自己的实际,不要盲从。这也是老生常谈的问题,比如线粒体基因比例,通常认为高线粒体基因比例代表细胞不好,具体的阈值要根据实际确定。其他的例如total_counts,也不能太高,如果同一批样本某些组中离群细胞的total_counts高,可能是因为双细胞,所以要cut一下。同样的,n_genes_by_counts,对于每个细胞检测到的基因数也需要控制,如果太低,那么这个细胞质量也不好,需要过滤。总之,需要结合自己的实际数据调整。这关系到后续的细胞分群注释。
adata = adata[
(adata.obs.pct_counts_mt < 20) &
(adata.obs.pct_counts_ribo < 40) &
(adata.obs.n_genes_by_counts < 6000) &
(adata.obs.total_counts >= 1000) &
(adata.obs.total_counts < 20000) &
(adata.obs.pct_counts_hb < 0.1), :].copy()
sc.pl.violin(
adata, #adata object
["n_genes_by_counts", "total_counts", "pct_counts_mt",'pct_counts_ribo', 'pct_counts_hb'], #adata.obs或者adata.var_names需要plot的值
jitter=0.2,#抖动点宽度
size=2,#点的大小
multi_panel=True
)