小罗碎碎念
想象一下,如果你去体检时,医生只告诉你"血压正常"或"血压异常",却不告诉你具体数值------你无法知道自己是接近正常上限还是严重超标,后续调理也没有精准方向。
在癌症病理诊断中,类似的"粗糙判断"已经存在了多年:传统的深度学习技术分析病理切片时,总是把像"同源重组缺陷(HRD)"这样的关键分子标志物,简单归为"有"或"无"两类。
但实际上,这些标志物更像血压、血糖,是连续变化的"刻度值",强行分类会丢失大量关键信息,就像用"冷热"描述体温,却忽略了37℃和39℃的本质差异。
数字病理的出现本是为了突破传统显微镜的局限:通过将病理切片扫描成亿像素级的全切片图像(Whole Slide Image, WSI),再用深度学习挖掘其中的分子信息。
比如,医生曾希望AI能从WSI中直接"看"出肿瘤是否存在特定基因突变,辅助精准用药。但问题在于,绝大多数已有的AI模型都在用"分类"思维处理问题------它们需要先把连续的生物标志物(如HRD分数、基因表达量)切成"阳性/阴性"的二元标签,这个过程就像把连续的身高数据分成"高/矮",必然会丢失中间的渐变信息。
而正是这些丢失的信息,可能藏着判断肿瘤恶性程度、患者对药物敏感性的关键线索。
直到近期,发表在《Nature Communications》的一项研究提出了回归型深度学习(Regression-based Deep Learning) 方案,特别是其中的CAMIL回归模型(Contrastively-Clustered Attention-based Multiple Instance Learning Regression) ,试图打破这一局限。

它不再强行给生物标志物"贴标签",而是直接预测其连续数值,就像从"非黑即白"的判断,升级为"精准到小数点后"的测量。
这一改变,不仅让AI对分子标志物的预测更准确,还能为癌症患者的预后判断提供更可靠的依据。
基于对比聚类注意力的多实例学习回归方法(CAMIL regression)
该方法整合自监督学习(SSL)与注意力机制的弱监督回归框架,以11671张涵盖9种癌症类型的病理切片为研究对象,直接预测连续型生物标志物,核心突破在于避免将连续值转化为离散分类标签;
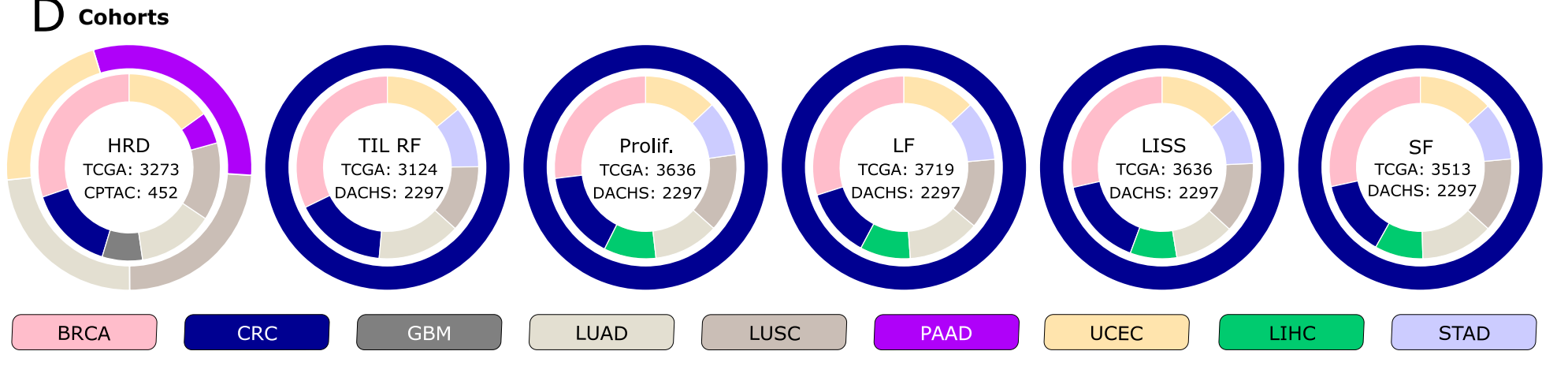
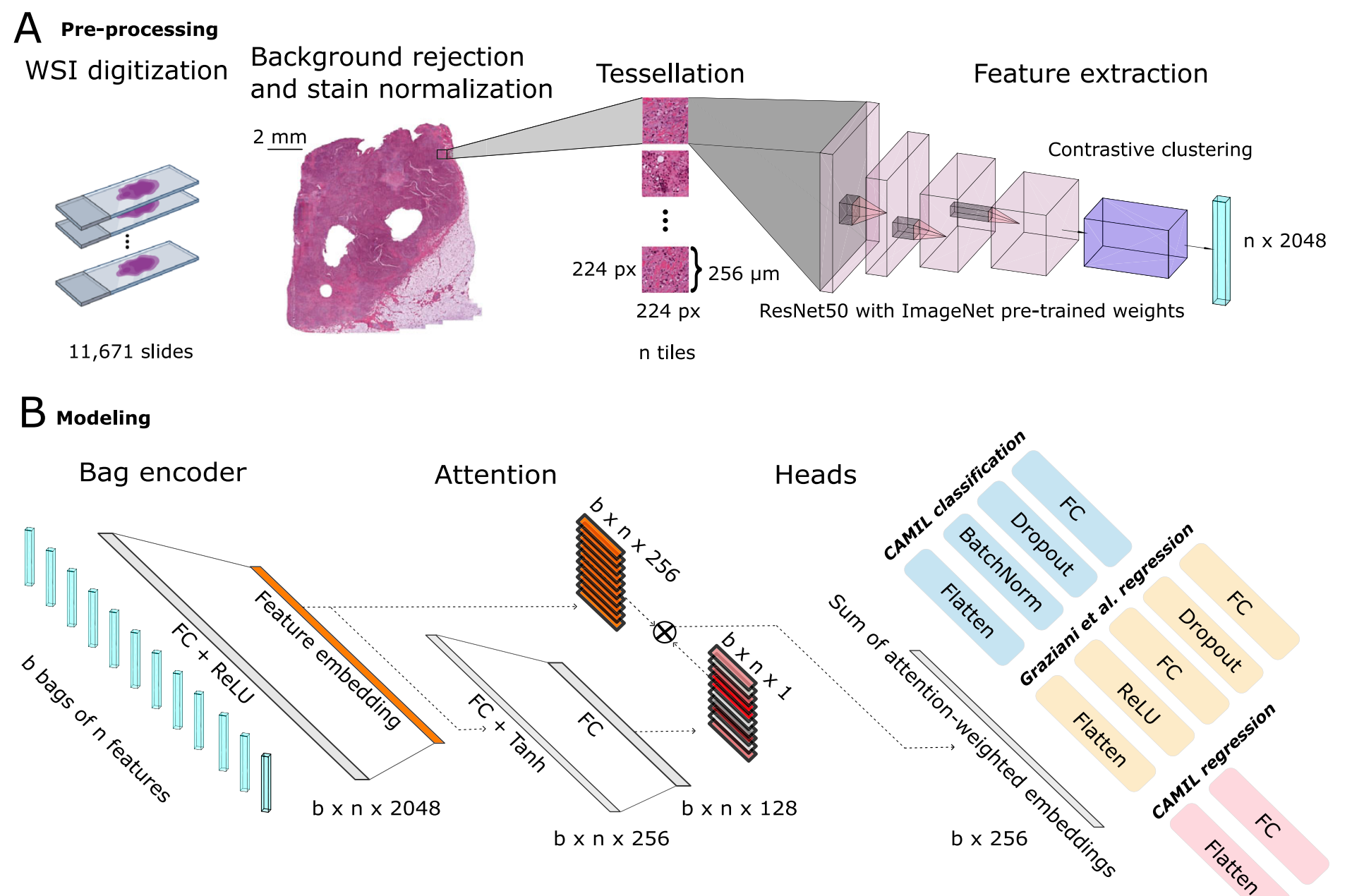
同时通过ResNet50预训练模型结合RetCCL对比聚类进行特征提取,再经attMIL模型实现患者水平的预测聚合,有效保留生物标志物的连续信息以提升预测精度。
 在性能验证方面,研究以同源重组缺陷(HRD)这一泛癌临床生物标志物及肿瘤微环境关键生物学过程标志物为核心验证对象,通过TCGA内部队列5折交叉验证与CPTAC外部队列验证,证实CAMIL regression在预测准确性与稳定性上显著优于传统分类方法及现有回归方法。
在性能验证方面,研究以同源重组缺陷(HRD)这一泛癌临床生物标志物及肿瘤微环境关键生物学过程标志物为核心验证对象,通过TCGA内部队列5折交叉验证与CPTAC外部队列验证,证实CAMIL regression在预测准确性与稳定性上显著优于传统分类方法及现有回归方法。
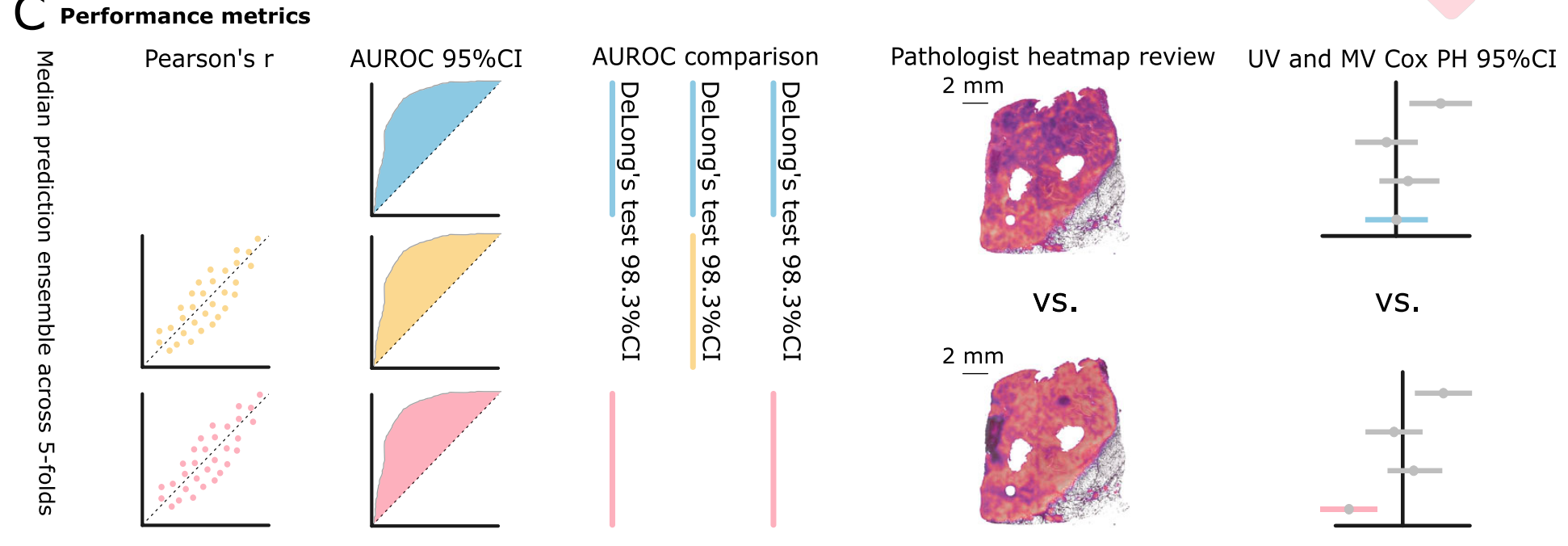
研究还进一步验证了CAMIL regression在临床预后预测中的价值,在包含2297例结直肠癌患者的DACHS外部队列中,将基于乳腺癌TCGA队列训练的模型迁移应用于结直肠癌生存分析,发现CAMIL regression预测的生物标志物在单变量与多变量Cox比例风险模型中,能更有效实现风险分层。
这一结果证实了该回归方法的泛化能力与临床转化潜力,为医学AI在病理生物标志物分析及预后预测中的应用提供了新的有效框架。
加入团队
罗小罗团队是一支以国内外硕博为主的科研团队,覆盖影像组学、病理组学以及基因组学等医学AI主流研究领域。
团队目前拥有7名副教授,60+硕博,多名成员以第一作者身份在Nature(2篇)、Nature Communications、Advanced Science以及Radiology等顶级期刊发表过论文。
如果想要加入我们团队,欢迎投递个人简历到团队邮箱:lxltx2025@163.com
医学AI交流群
目前小罗全平台关注量120,000+,交流群总成员3000+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
影像组学培训课程(团队自营)
11月团队将在【北京】、【上海】和【广州】三个城市开展线下培训课程,可以选择任意一个城市参加。
一、从"分类"到"回归":CAMIL模型如何让病理分析更精准?
要理解CAMIL回归的突破,我们得先搞清楚它解决了传统方法的两个核心问题:信息丢失(二值化导致)和注意力偏差(无法聚焦关键组织区域)。
它的创新逻辑很简单:
- 先通过自监督学习让模型"读懂"病理切片的基础特征
- 再用注意力机制聚焦关键区域
- 最后直接输出连续的生物标志物数值------全程不做任何"一刀切"的分类处理
我们可以把整个技术流程想象成"侦探分析犯罪现场":
- 先整理现场(预处理切片)
- 再提取关键线索(特征提取)
- 最后聚焦核心证据做判断(注意力回归)
预处理------给病理切片"整理现场"
病理切片的质量受染色、扫描设备影响很大,就像同一张照片,有人拍得亮、有人拍得暗,直接分析会出错。
CAMIL的第一步就是"标准化":
切片分割
把亿像素的WSI切成224×224像素的小组织块(Tile),每个小块对应实际组织的256微米,相当于把一幅巨画切成明信片大小,方便逐一分析;
去"干扰项"
用边缘检测技术去掉空白背景、模糊组织(如坏死区域),就像侦探排除无关的现场杂物;
染色归一化
统一不同切片的染色深浅,避免"有的切片染得红、有的染得紫"导致模型误判,这一步类似把所有照片调成统一的亮度和色调。
特征提取------用"专业工具"找线索
传统模型直接用预训练的图像识别模型(如ResNet)提取特征,但CAMIL多做了一步自监督学习(Self-Supervised Learning, SSL) :
它先让模型在3.2万张不同癌症的病理切片上"自学"------不用人工标注,模型自己找切片里的共性特征(比如"癌细胞的细胞核更密集""免疫细胞的形状特殊"),就像侦探先研究上千个犯罪现场,总结出"小偷通常会留下指纹"这样的规律;
然后用经过自监督训练的ResNet50模型,给每个小组织块提取2048维的"特征向量"------可以理解为给每个组织块发一张"身份卡",上面记录了它的细胞类型、结构特点等关键信息。
这一步的巧妙之处在于:自监督学习让模型对病理组织的理解更"深刻",不用依赖大量人工标注的数据集,解决了病理标注成本高、样本少的痛点。
注意力回归------聚焦核心证据,算"精确分数"
最关键的突破在这里------病理切片中,只有少数区域(如癌细胞聚集区)对预测分子标志物有用,其他区域(如脂肪、结缔组织)是干扰项。
CAMIL用注意力多实例学习(attMIL) 解决这个问题:
- 它把每个患者的所有组织块特征看作"一叠报告",注意力机制就像主管批改报告------会给关键报告(如癌细胞区域)打高分,给无关报告(如脂肪组织)打低分;
- 然后通过多层神经网络,把这些"加权后的特征"聚合起来,直接输出连续的生物标志物数值(如HRD分数0-103,免疫细胞比例0-100%),而不是"阳性/阴性"标签。
为了让回归更准确,CAMIL还做了两个优化:
- 用Adam优化器避免模型"钻牛角尖"(比如传统优化器容易让预测值偏向平均值)
- 用Balanced MSE损失函数处理"癌症样本中阳性少、阴性多"的不平衡问题
二、CAMIL回归到底比传统方法好在哪?
研究团队用11671张来自9种癌症的病理切片,从"预测准确性"和"临床实用性"两个维度,对比了CAMIL回归与传统分类模型、其他回归模型(如Graziani等人的方法)的效果。
预测HRD------泛癌标志物的"精准度测试"
HRD(同源重组缺陷)是判断肿瘤是否对铂类化疗药、PARP抑制剂敏感的关键指标,它的真实值是0-103的连续分数,传统方法会用"≥42为阳性"切成两类。
研究团队用TCGA(癌症基因组图谱)的7种癌症数据训练模型,再用CPTAC(临床蛋白质组肿瘤分析联盟)的外部数据验证:
准确性更高
在子宫内膜癌(UCEC)中,CAMIL回归的AUROC(衡量区分能力的指标,越接近1越好)达到0.96,比传统分类模型的0.98虽略低,但关键是------CAMIL回归能把HRD阳性和阴性患者的预测分数分得更开(见图2G、H)。
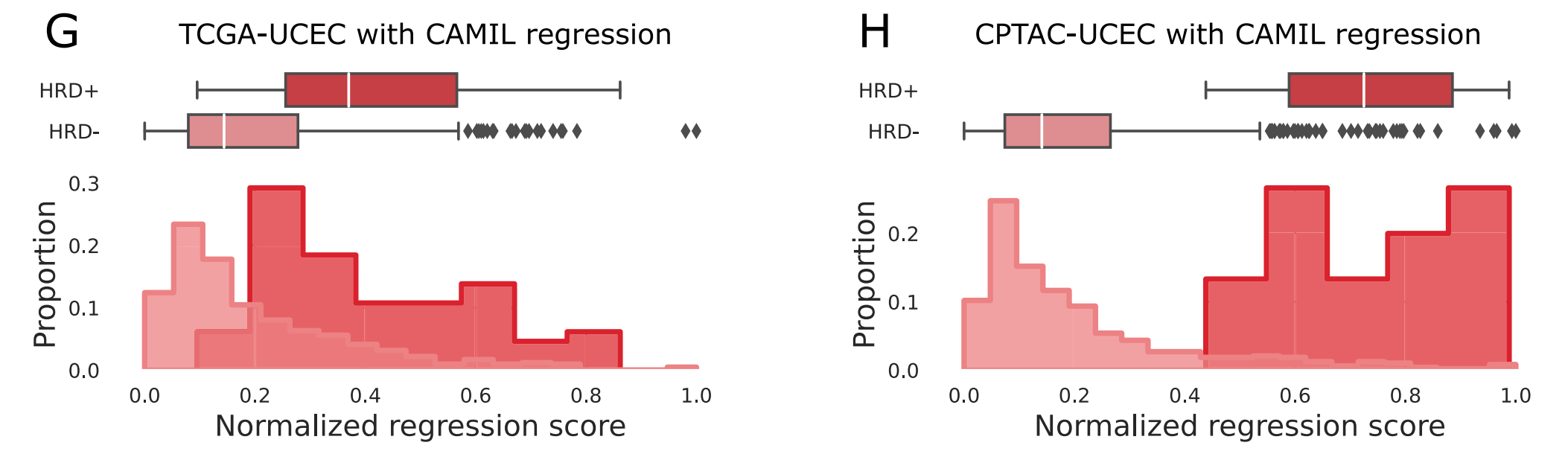
传统分类模型的分数分布有重叠,就像"把37℃和38℃都归为'发烧',但分不清严重程度",而CAMIL回归能精确到具体分数,医生能更清楚患者的HRD水平;
稳定性更强
在5折交叉验证中,CAMIL回归的性能波动更小,比如在乳腺癌(BRCA)中,它的AUROC波动范围比传统分类小15%,说明模型不容易受数据差异影响;
外部验证更可靠
在CPTAC的肺癌(LUAD)数据中,CAMIL回归的AUROC达到0.81,比Graziani回归的0.72高12%,证明它在不同医院、不同扫描设备的切片上都能用。
结直肠癌预后------帮医生判断患者生存风险
临床中,医生需要通过生物标志物判断患者的生存风险(如"免疫细胞多的患者可能活得更久")。
研究团队用2297名结直肠癌患者(DACHS队列)的10年随访数据,测试模型的预后价值:
分层更精准
传统分类模型只能在2个生物标志物(如免疫细胞比例、增殖水平)上区分患者的生存风险,而CAMIL回归能在3个标志物上显著分层,且风险差异更明显。
比如在"白细胞分数(LF)"这个指标上,CAMIL回归预测的高LF患者,比低LF患者的死亡风险低76%(HR=0.24),而传统分类模型只能算出低50%(HR=0.5);
多变量分析更稳定
当加入年龄、性别、肿瘤分期等其他因素后,传统分类模型只有1个标志物仍能预测预后,而CAMIL回归有2个(LF和LISS),说明它的预测结果受其他因素干扰更小,对医生更有参考价值;
反观Graziani回归模型,由于预测值差异太小,甚至无法让生存分析模型收敛------就像"所有人的风险分数都差不多,医生没法区分谁更危险",这也证明了CAMIL回归的优越性。
三、从实验室到临床:这个技术能改变什么?
CAMIL回归的价值,不止是"比传统方法准一点",更在于它能解锁病理切片中"被忽略的信息",推动数字病理从"辅助诊断"走向"精准预测"。
当下:解决临床痛点
拯救"存量切片"
全球医院积累了上亿张病理切片,但大多数没有分子标志物数据(如HRD、基因表达量)。
用CAMIL回归,能直接从这些"老切片"中预测分子信息,不用再给患者做基因检测------既节省成本(一次基因检测数千元),又能让老病例数据"复活",用于研究;
优化治疗方案
比如HRD分数,传统分类只能判断"能用PARP抑制剂"或"不能用",而CAMIL回归能给出具体分数------医生可以根据分数调整药量,避免"分数刚达标就用药"导致的副作用,或"分数接近达标却不用药"的错过机会。
未来:打开更多可能
跨癌种预测
目前CAMIL已在9种癌症上验证,未来有望扩展到更多癌症类型,甚至实现"一张切片预测多个分子标志物",比如同时算出HRD、TMB(肿瘤突变负荷)、MSI(微卫星不稳定),为医生提供"一站式"信息;
多组学重建
病理切片是"形态学数据",分子标志物是"分子数据",CAMIL回归搭建了两者的桥梁。
未来可能结合基因组、蛋白质组数据,用病理切片"反向重建"患者的多组学信息,让精准医疗更易落地;
推动AI病理标准化
由于CAMIL回归的稳定性和可解释性(注意力热图能显示模型关注的区域,医生可验证),它可能成为AI病理的"新标准",解决目前不同模型"各说各话"的问题。
就像血压计从"有无高血压"的定性判断,升级为"精确到1mmHg"的定量测量,CAMIL回归也让病理分析从"非黑即白"走向"精准刻度"。
它的意义不仅在于技术突破,更在于让病理切片这一"最传统的诊断材料",焕发出"预测分子信息"的新价值------未来,或许医生只要在显微镜下扫一眼切片,AI就能同步给出患者的分子标志物分数、推荐的治疗方案,让精准医疗真正走进每个医院。
参考文献
本文基于Omar S. M. El Nahhas等人发表于《Nature Communications》(2024, 15:1253)的研究成果,原文标题为"Regression-based Deep-Learning predicts molecular biomarkers from pathology slides",可通过DOI:10.1038/s41467-024-45589-1查阅。
研究中涉及的CAMIL模型代码、预处理流程已开源,可在KatherLab的GitHub仓库(如https://github.com/KatherLab/marugoto)获取。