StoL:像搭乐高一样用扩散模型构建大分子 3D 构象
目录
- MergeDNA 利用动态分词策略适配基因组复杂性,在多组学任务中超越现有 SOTA 模型。
- 这种新型混合深度学习框架引入原子杂化状态,并结合传统分子描述符,成功突破了单一 GNN 模型难以捕捉全局物化性质的瓶颈。
- PepBFN 利用黎曼 - 欧几里得贝叶斯流网络,在全连续参数空间统一建模序列与结构,解决侧链旋转异构体的多模态分布难题。
- SSRGNet 结合序列语言模型与 3D 空间图网络,利用多重关系边和并行融合策略,提升了蛋白质二级结构预测的精度。
- StoL 通过将大分子拆解为片段,利用扩散模型分别生成 3D 结构后组装,巧妙解决了大分子构象生成中训练数据匮乏的难题。
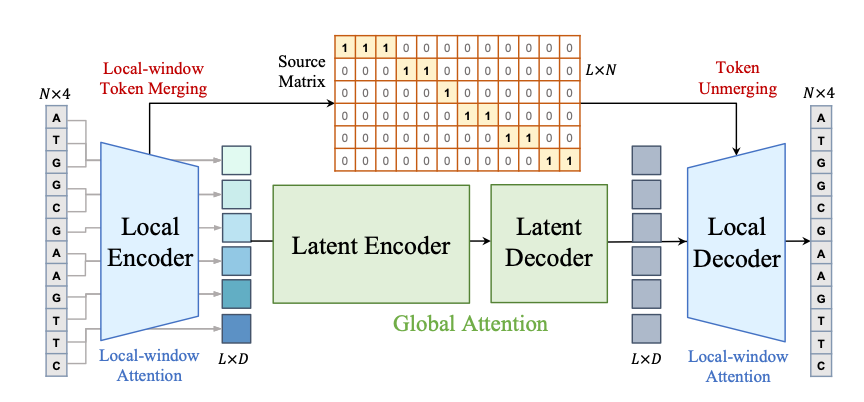
1. MergeDNA:基因组建模的动态分词突破
现有的 DNA 大语言模型(DNA Foundation Models)常因处理方式深受人类语言模型影响而陷入困境。主流的 k-mer 或字节对编码(BPE)难以应对基因组极不均匀的信息密度:高密度编码区哪怕一个碱基的变动都至关重要,而大段重复序列则包含极低信息量。用同一把尺子衡量这两类区域并不合理。
MergeDNA 引入了「变焦镜头」式的分层架构。它利用局部编码器(Local Encoder)配合可微分的 Token 合并(Token Merging)技术解决这一难题。
遇到高度重复、低信息量序列,模型自动合并 Token,实现「一目十行」;面对复杂的启动子或外显子区域,则保留细粒度 Token,做到「逐字精读」。这种动态调整既能捕捉短程基序(Motifs),又能高效处理长程依赖关系。
训练该架构依赖两个关键预训练任务:
- 合并 Token 重建(Merged Token Reconstruction):模型需还原合并后的 Token,迫使其理解被压缩区域的上下文。
- 自适应掩码建模(Adaptive Masked Token Modeling):进阶版掩码语言模型,自适应过滤重要 Token 进行掩码预测。
MergeDNA 在多个 DNA 基准测试和多组学任务中达成 SOTA(State-of-the-Art)。在跨物种泛化、RNA 剪接位点预测及零样本蛋白适应性预测(Zero-shot protein fitness prediction)等硬核任务上,表现优于传统大规模模型。
对于计算辅助药物发现(CADD)和合成生物学,这提供了更高分辨率的工具。模型超越了机械的碱基概率统计,真正开始识别基因组中的信号与噪音。
📜Title: MergeDNA: Context-aware Genome Modeling with Dynamic Tokenization through Token Merging
🌐Paper: https://arxiv.org/abs/2511.14806v1
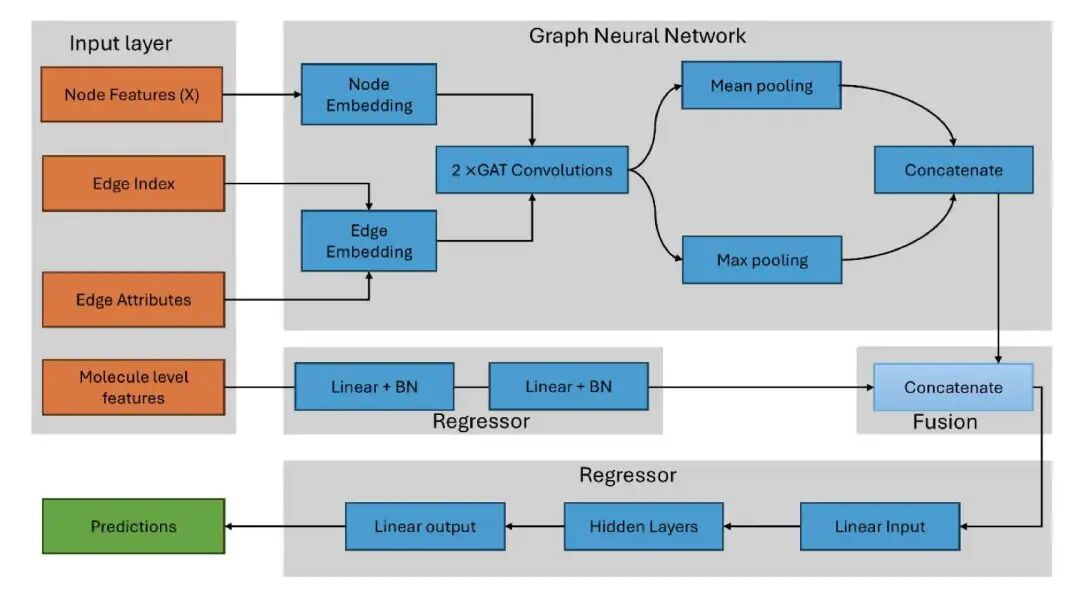
2. 原子杂化赋能 GNN:药物靶点预测精准度大增
给 AI 装上「化学家的眼睛」
计算辅助药物发现常将分子简化为二维图,原子仅作为节点,忽略了更深层的化学性质。但在药物化学家眼中,苯环上的碳(sp2 杂化)与连着四个单键的碳(sp3 杂化)天壤之别:它们的空间占据、电子云分布及反应活性截然不同。
该研究开发了一种基于原子杂化(Atomic Hybridization)的图神经网络(GNN)模型。模型不再局限于基础原子类型,而是深入电子构型和空间取向层面。这种处理方式让算法能像化学家一样,从电子层面感知分子的局部结构。
新老结合:GNN 与分子描述符的联姻
纯 GNN 模型往往过于关注局部连接,容易忽略脂水分配系数 logP 或分子量等整体性质。为解决此问题,研究者设计了一种混合架构:一条支路通过 GNN 处理局部结构,另一条专门处理经典分子描述符。
这如同评估药物分子时,既用显微镜观察官能团细节,又参考宏观物化指标。这种协同效应提升了预测精度,并保留了可解释性------我们可以看到哪些物理化学性质在发挥作用。
数据与结果:硬碰硬的提升
研究人员利用 9 个生物靶点、14,316 个化合物进行训练。测试集 R² 达到 0.87,性能较既往方法提升 6% 至 42%。在激酶、核受体等高成药性靶点上,模型展现出优异的 IC₅₀ 预测能力。
多靶点联合训练是另一大亮点。针对新靶点数据匮乏的痛点,该模型允许知识在不同蛋白家族间迁移。在数据丰富靶点上学到的规律,可辅助预测数据稀缺靶点,这对仅有少量活性数据的孤儿受体极具实用价值。
若后续引入 3D 构象信息,或利用大规模预训练模型进行微调,该框架的潜力将得到进一步释放。
📜Title: Graph Neural Networks Model Based on Atomic Hybridization for Predicting Drug Targets
🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.19.689219v1
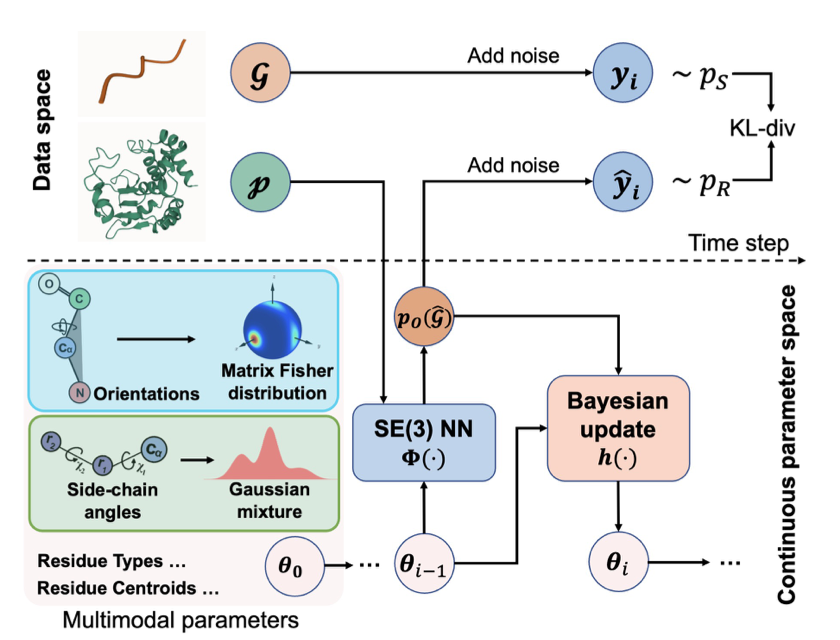
3. PepBFN:贝叶斯流重塑全原子多肽设计
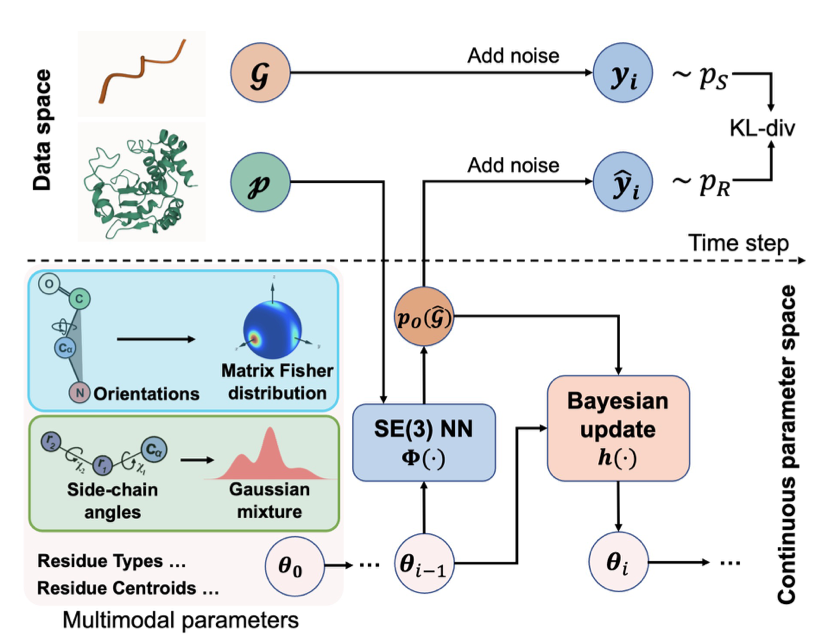
多肽设计长期面临一个痛点:离散的氨基酸序列与连续的三维结构坐标难以协同。PepBFN 引入贝叶斯流网络(Bayesian Flow Networks, BFNs),用一种数学上优雅的方式尝试解决这一难题。
全连续参数空间
传统扩散模型处理离散序列时涉及加噪去噪,难免损失信息。PepBFN 将序列和结构统一置于全连续参数空间。这种处理方式类似将数字信号转化为模拟信号,在捕捉细微变化时具备离散模型无法比拟的平滑度与连贯性。
侧链动态捕捉
氨基酸侧链并非只有固定姿态,而是存在旋转异构体(rotamers),类似开关的多个档位。传统模型常假设侧链服从单峰高斯分布,这违背物理规律。PepBFN 引入高斯混合模型(Gaussian mixture-based)的贝叶斯流,准确识别侧链在空间中的多个潜在稳定状态,而非强行取平均值。这种对多模态(multimodal)特性的捕捉提升了侧链预测精度。
SO(3) 流形几何归位
蛋白质骨架取向(Orientation)若用欧几里得几何计算,极易产生偏差且需频繁修正。该模型直接在 SO(3) 流形 上工作,采用基于 Matrix Fisher 分布的黎曼流。这种专门描述旋转的数学语言让贝叶斯更新易于计算,残基方向的变化在数学上自然连续,无需额外几何约束防止结构崩塌。
实战表现
理论优势转化为实际性能。在侧链包装(side-chain packing)、反向折叠(reverse folding)及高难度的结合体设计(binder design)任务上,PepBFN 刷新了现有最佳成绩。将旋转异构的多样性、几何空间的非欧几里得性等生物物理特性正确映射到数学模型中,计算辅助药物设计的精度便跃升新台阶。
📜Title: Full-Atom Peptide Design via Riemannian--Euclidean Bayesian Flow Networks
🌐Paper: https://arxiv.org/abs/2511.14516
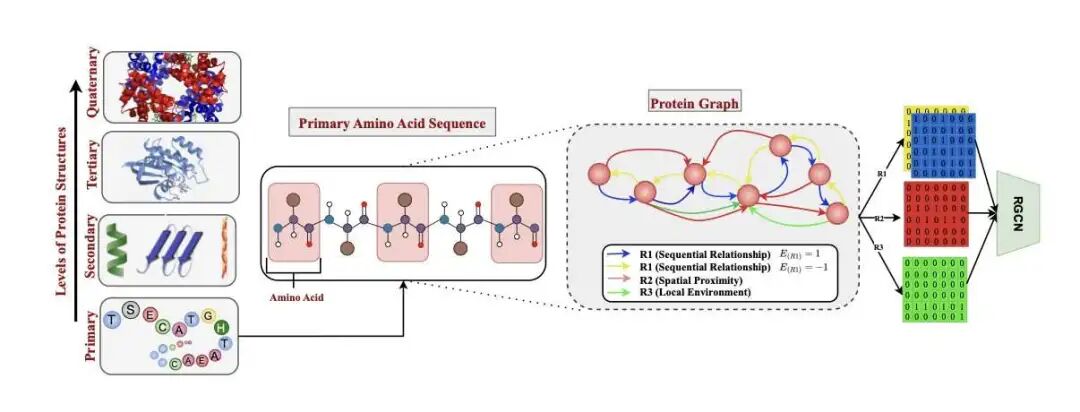
4. SSRGNet:3D 图神经网络与大语言模型联手预测蛋白质结构
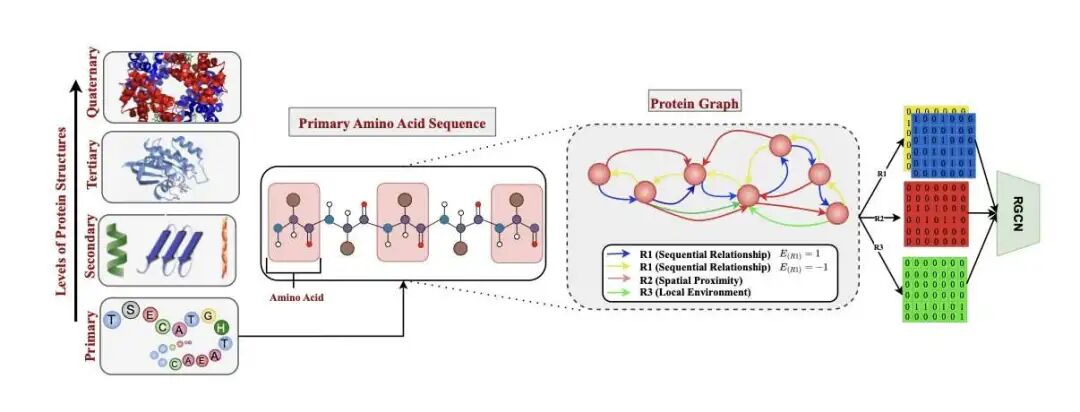
- 双重编码机制:使用 DistilProtBert 处理序列信息,结合关系型图卷积网络(R-GCN)处理 3D 结构特征。
- 多维关系建模:构建包含序列、空间和局部环境三种边缘类型的蛋白质图,捕捉复杂折叠模式。
- 并行融合优势:并行融合(Parallel Fusion)能比串行或交叉融合更有效地保留多模态信息的互补性。
- 性能验证:在 CB513、TS115 和 CASP12 等基准数据集上,准确率超越现有主流方法。
蛋白质结构预测领域正在融合自然语言处理(NLP)的大语言模型(LLM)与处理几何结构的图神经网络(GNN)。SSRGNet 展示了将「阅读」氨基酸序列与「观察」3D 拓扑结构结合的成果。
兼顾序列与形状
蛋白质本质是空间结构,仅关注序列会丢失关键的空间信息。SSRGNet 利用 DistilProtBert 提取序列中的上下文语义,同时构建 3D 蛋白质残基图,通过图神经网络捕捉空间几何特征。
三种「关系」定义残基
模型通过三种不同类型的「边(Edge)」描述残基间关系,而非简单连接相邻残基:
- 序列边(Sequential):关注前后相邻的氨基酸。
- 空间边(Spatial):连接序列距离远但在 3D 空间折叠后紧邻的氨基酸,捕捉长程相互作用。
- 局部环境边(Local Environment):描述残基周围微环境特征。
借助关系感知消息传递(Relation-Aware Message Passing),模型能综合分析氨基酸的身份、位置及其邻近环境的影响。
并行融合策略
面对序列与结构两类数据,并行融合(Parallel Fusion)效果优于串行或交叉融合。这种方式允许独立处理序列与结构特征,最后综合判断,最大程度保留了两种模态的互补信息。
结果与应用
CB513 和 CASP12 等数据集的测试结果验证了模型的有效性。消融实验证实,三种关系边对于模型性能缺一不可。后续计划整合更多外部结构数据集进行预训练。对于药物设计或酶工程领域,这种能精细解读蛋白质局部几何结构的模型具有重要实用价值。
📜Title: Protein Secondary Structure Prediction Using 3D Graphs and Relation-Aware Message Passing Transformers
🌐Paper: https://arxiv.org/abs/2511.13685v1
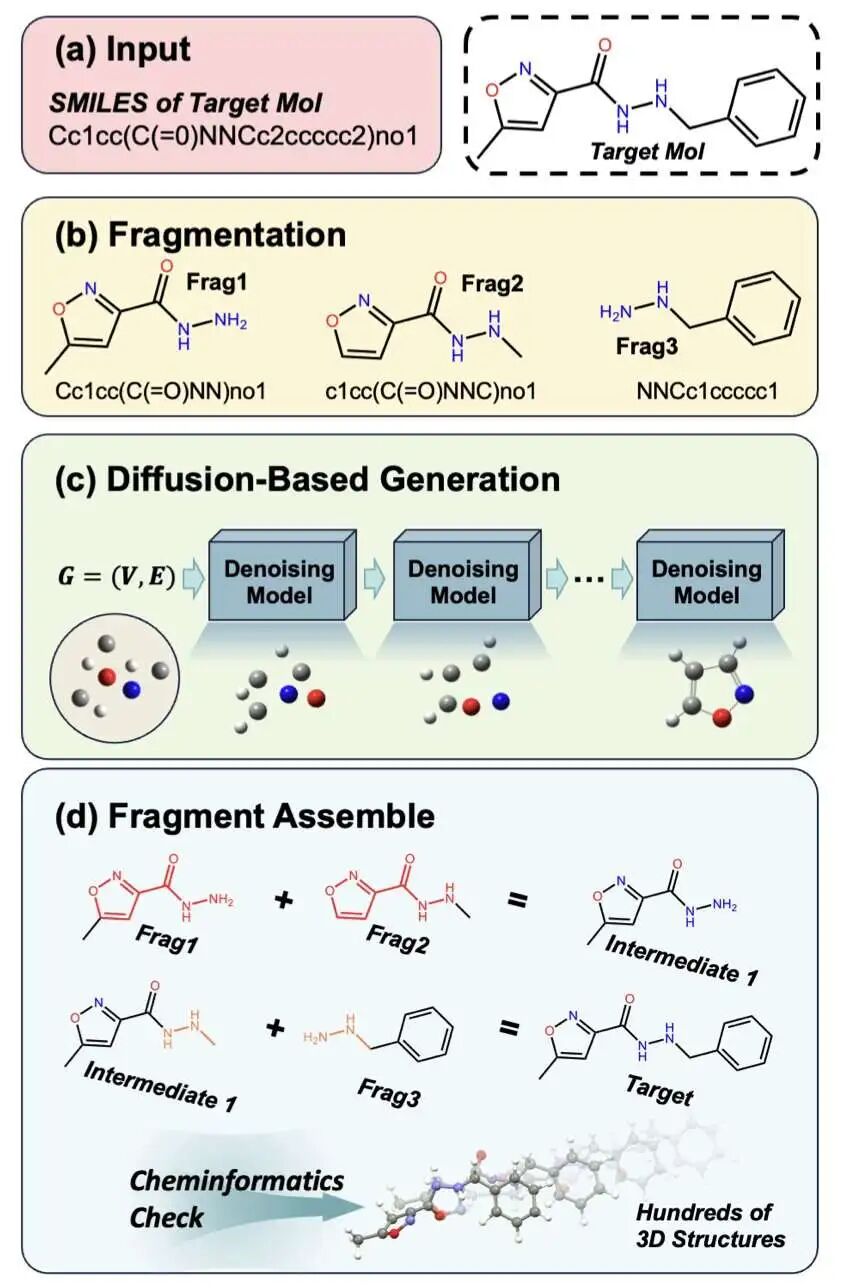
5. StoL:像搭乐高一样用扩散模型构建大分子 3D 构象
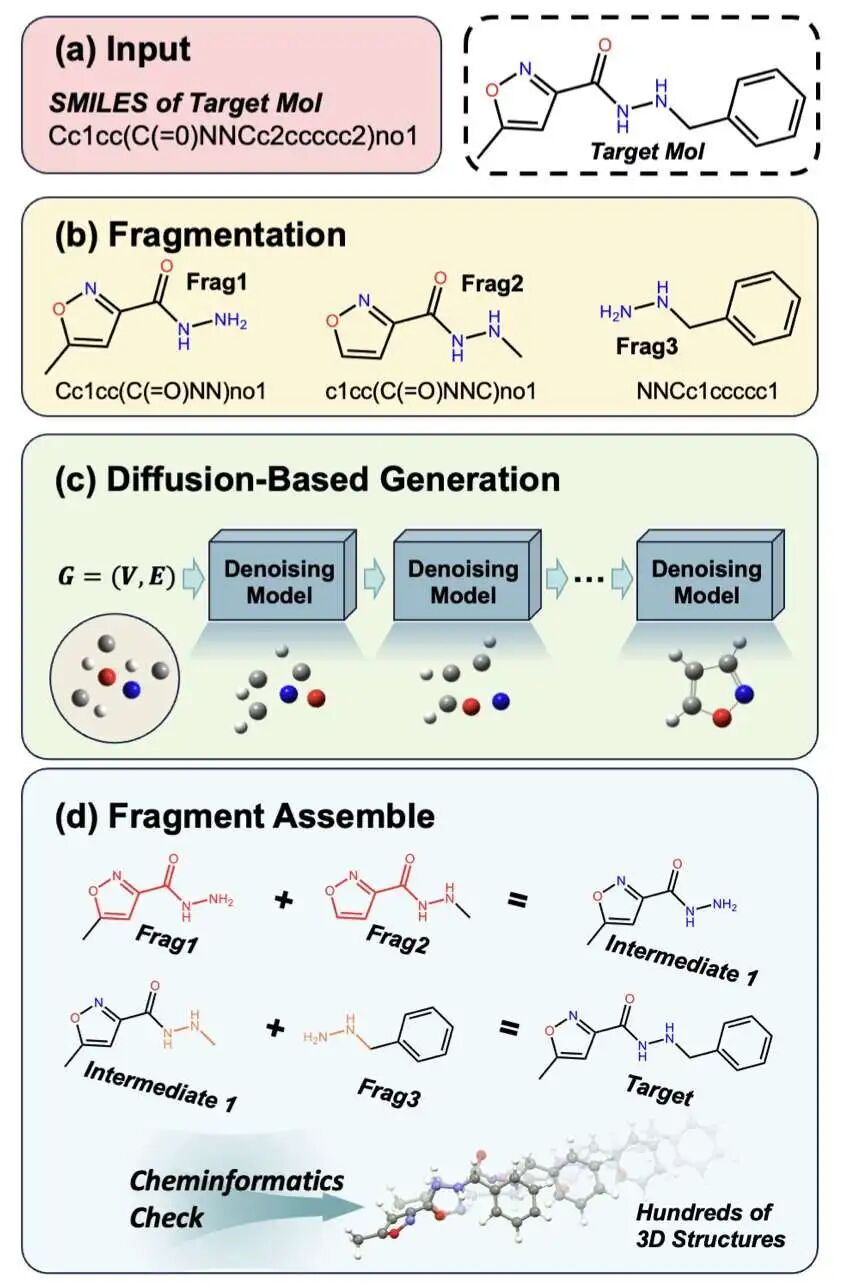
药物研发始于精准的分子 3D 构象。小分子领域数据充足,处理起来得心应手;但大分子的高质量 3D 训练数据极度匮乏,直接使用端到端深度学习模型效果往往不尽人意。
这项名为 StoL (Small-to-Large) 的研究另辟蹊径:既然大分子数据不够,那就将其拆解,利用小分子领域的优势来解决问题。
像搭积木一样组装分子
StoL 的工作逻辑直观且高效。它将复杂的长链分子沿可旋转单键切开,分解为化学上合理的刚性片段。
这一操作将预测整个大分子复杂折叠的难题,转化为多个简单的局部 3D 生成任务。现有的扩散模型在小分子数据上训练充分,能轻松处理这些碎片。系统随后依据连接点信息,将生成好的 3D 碎片重新组装。这种方法有效避开了大分子数据不足的障碍。
注入「化学常识」
仅靠数据驱动容易导致模型犯低级错误,例如将本应平面的芳香环扭曲。StoL 为此设计了「化学增强」训练阶段。
模型首先通过大量数据学习分子的总体形态,随后进入第二阶段,接受物理化学规则的专项训练。这种策略强制模型遵守键长、键角和平面性等基本原则。最终生成的结构不仅几何形态合理,能量稳定性也大幅提升。
超越 RDKit 的搜索能力
常用的 RDKit 工具虽然速度快,但容易陷入局部最优解,难以找到能量最低的天然状态。
StoL 在这方面表现出色。通过计算化学的金标准------密度泛函理论 (DFT) 对比验证,StoL 生成的构象能量往往更低。这意味着它能探索更广阔的化学空间,发现传统方法遗漏且在生物体内真实存在的分子姿态。对于设计需要精确契合蛋白口袋的药物,这一优势至关重要。
局限与启示
StoL 并非完美,在处理难以简单切割的复杂稠环系统时仍显吃力。但这依然是计算化学领域的一次巧妙尝试,它展示了解决复杂问题的有效路径:将其拆解为我们已经能解决的简单问题。
📜Title: Chemistry-Enhanced Diffusion-Based Framework for Small-to-Large Molecular Conformation Generation