#!/usr/bin/env Rscript
# Seurat V5 + Harmony 批次校正流程 - 支持CSV和10X格式
# 使用: Rscript harmony_integration.R --samples sample.txt --output results
suppressPackageStartupMessages({
library(Seurat)
library(ggplot2)
library(patchwork)
library(dplyr)
library(argparse)
library(future)
library(harmony)
})
# 创建参数解析器
parser <- ArgumentParser(description = 'Seurat V5 + Harmony 批次校正流程 - 支持CSV和10X格式')
# 添加参数
parser$add_argument('--samples', type='character', required=TRUE,
help='样本信息文件,四列:样本名、数据路径、数据类型(10X/csv)、分组信息')
parser$add_argument('--output', type='character', default='harmony_results',
help='输出目录路径')
parser$add_argument('--min_cells', type='integer', default=3,
help='基因至少在多少细胞中表达')
parser$add_argument('--min_features', type='integer', default=200,
help='细胞至少检测到多少基因')
parser$add_argument('--max_features', type='integer', default=5000,
help='细胞最多检测到多少基因')
parser$add_argument('--max_mt', type='double', default=20,
help='最大线粒体基因百分比')
parser$add_argument('--max_rb', type='double', default=50,
help='最大核糖体基因百分比')
parser$add_argument('--nfeatures', type='integer', default=2000,
help='高变基因数量')
parser$add_argument('--npcs', type='integer', default=50,
help='PCA计算维度')
parser$add_argument('--ndims', type='integer', default=30,
help='用于Harmony和UMAP的维度')
parser$add_argument('--resolution', type='double', default=0.8,
help='聚类分辨率')
parser$add_argument('--harmony_vars', type='character', default='sample',
help='Harmony校正的变量,逗号分隔')
parser$add_argument('--harmony_max_iter', type='integer', default=20,
help='Harmony最大迭代次数')
parser$add_argument('--threads', type='integer', default=4,
help='并行线程数')
parser$add_argument('--csv_sep', type='character', default=',',
choices=c(',', ';', '\t', ' '),
help='CSV文件分隔符')
parser$add_argument('--csv_has_rownames', action='store_true',
help='CSV文件是否有行名(基因名)')
parser$add_argument('--csv_has_colnames', action='store_true',
help='CSV文件是否有列名(细胞名)')
# 解析参数
args <- parser$parse_args()
# 创建输出目录
if (!dir.exists(args$output)) {
dir.create(args$output, recursive = TRUE)
}
# 设置并行
plan("multicore", workers = args$threads)
options(future.globals.maxSize = 20 * 1024^3) # 10GB
# 解析Harmony变量
harmony_vars <- unlist(strsplit(args$harmony_vars, ","))
# 函数:根据数据类型读取数据
read_sc_data <- function(data_path, data_type, sample_name) {
cat(paste(" 读取", data_type, "格式数据...\n"))
if (data_type == "10X") {
# 10X Genomics格式
if (!dir.exists(data_path)) {
stop(paste("10X数据目录不存在:", data_path))
}
# 读取10X数据
data <- Read10X(data.dir = data_path)
cat(paste(" 成功读取10X数据,矩阵维度:", dim(data)[1], "×", dim(data)[2], "\n"))
} else if (data_type == "csv") {
# CSV格式 - 直接使用read.csv
if (!file.exists(data_path)) {
stop(paste("CSV文件不存在:", data_path))
}
cat(paste(" 读取CSV文件,分隔符:", args$csv_sep, "\n"))
# 使用read.csv读取
data <- read.csv(
data_path,header = T,row.names = 1,
check.names = FALSE,
stringsAsFactors = FALSE
)
# 转换为矩阵
data <- as.matrix(data)
cat(paste(" 成功读取CSV数据,矩阵维度:", dim(data)[1], "×", dim(data)[2], "\n"))
} else {
stop(paste("不支持的数据类型:", data_type, "。支持的类型: 10X, csv"))
}
return(data)
}
# 记录参数
cat("========================================\n")
cat("Seurat V5 + Harmony 批次校正流程参数设置:\n")
cat("========================================\n")
cat(paste("样本文件:", args$samples, "\n"))
cat(paste("输出目录:", args$output, "\n"))
cat(paste("数据格式支持: 10X, csv\n"))
cat(paste("CSV分隔符:", args$csv_sep, "\n"))
cat(paste("CSV有行名:", args$csv_has_rownames, "\n"))
cat(paste("CSV有列名:", args$csv_has_colnames, "\n"))
cat(paste("Harmony校正变量:", paste(harmony_vars, collapse=", "), "\n"))
cat(paste("最小细胞数:", args$min_cells, "\n"))
cat(paste("最小基因数:", args$min_features, "\n"))
cat(paste("最大基因数:", args$max_features, "\n"))
cat(paste("最大线粒体百分比:", args$max_mt, "%\n"))
cat(paste("最大核糖体百分比:", args$max_rb, "%\n"))
cat(paste("高变基因数:", args$nfeatures, "\n"))
cat(paste("PCA维度:", args$npcs, "\n"))
cat(paste("Harmony/UMAP维度:", args$ndims, "\n"))
cat(paste("聚类分辨率:", args$resolution, "\n"))
cat(paste("Harmony最大迭代:", args$harmony_max_iter, "\n"))
cat(paste("并行线程数:", args$threads, "\n"))
cat("========================================\n\n")
# 1. 读取样本信息
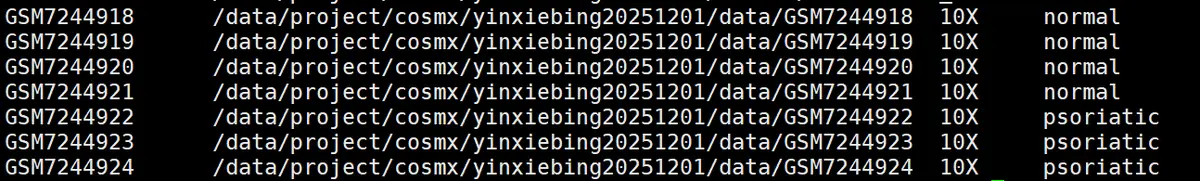
cat("1. 读取样本信息...\n")
samples_df <- read.table(args$samples, header = FALSE, sep = "\t", stringsAsFactors = FALSE,
col.names = c("sample_name", "data_path", "data_type", "group"))
# 验证数据类型
valid_types <- c("10X", "csv")
invalid_types <- setdiff(samples_df$data_type, valid_types)
if (length(invalid_types) > 0) {
stop(paste("无效的数据类型:", paste(invalid_types, collapse=", "), "。有效类型:", paste(valid_types, collapse=", ")))
}
cat(paste("找到", nrow(samples_df), "个样本:\n"))
for (i in 1:nrow(samples_df)) {
cat(paste(" ", samples_df$sample_name[i], ":",
samples_df$data_path[i], " [", samples_df$data_type[i], "] 分组:", samples_df$group[i], "\n"))
}
# 2. 加载和创建Seurat对象
cat("\n2. 加载单细胞数据...\n")
seurat_list <- list()
for (i in 1:nrow(samples_df)) {
sample_name <- samples_df$sample_name[i]
data_path <- samples_df$data_path[i]
data_type <- samples_df$data_type[i]
group_info <- samples_df$group[i]
cat(paste(" 加载样本:", sample_name, " [", data_type, "] 分组:", group_info, "\n"))
# 读取数据
data <- read_sc_data(
data_path = data_path,
data_type = data_type,
sample_name = sample_name
)
# 验证数据
if (is.null(data) || nrow(data) == 0 || ncol(data) == 0) {
stop(paste("样本", sample_name, "的数据为空或无效"))
}
# 确保基因名是字符型
if (is.null(rownames(data))) {
rownames(data) <- paste0("Gene_", 1:nrow(data))
cat(" 警告: 数据没有行名,已自动生成基因名\n")
}
# 确保细胞名是字符型且唯一
if (is.null(colnames(data))) {
colnames(data) <- paste0(sample_name, "_Cell_", 1:ncol(data))
} else {
# 添加样本名前缀确保细胞名唯一
colnames(data) <- paste0(sample_name, "_", colnames(data))
}
# 创建Seurat对象
seurat_obj <- CreateSeuratObject(
counts = data,
project = sample_name,
min.cells = args$min_cells,
min.features = args$min_features
)
# 添加样本信息、数据类型和分组信息
seurat_obj$sample <- sample_name
seurat_obj$data_type <- data_type
seurat_obj$group <- group_info
seurat_obj$orig.ident <- sample_name
# 添加到列表
seurat_list[[sample_name]] <- seurat_obj
cat(paste(" 细胞数:", ncol(seurat_obj), "基因数:", nrow(seurat_obj), "\n"))
# 清理内存
rm(data)
gc()
}
# 3. 合并所有样本
cat("\n3. 合并所有样本...\n")
if (length(seurat_list) == 1) {
seurat_obj <- seurat_list[[1]]
} else {
seurat_obj <- merge(
seurat_list[[1]],
y = seurat_list[-1],
add.cell.ids = samples_df$sample_name,
project = "merged_project"
)
}
cat(paste("合并后总细胞数:", ncol(seurat_obj), "\n"))
cat(paste("合并后总基因数:", nrow(seurat_obj), "\n"))
cat(paste("数据来源统计:\n"))
cat(paste(" 10X格式样本:", sum(samples_df$data_type == "10X"), "\n"))
cat(paste(" CSV格式样本:", sum(samples_df$data_type == "csv"), "\n"))
cat(paste("分组信息统计:\n"))
group_counts <- table(samples_df$group)
for (group_name in names(group_counts)) {
cat(paste(" ", group_name, ":", group_counts[group_name], "个样本\n"))
}
# 保存原始数据
saveRDS(seurat_obj, file.path(args$output, "1_merged_raw.rds"))
cat(paste("原始数据已保存到:", file.path(args$output, "1_merged_raw.rds"), "\n"))
# 4. 计算质量控制指标
cat("\n4. 计算质量控制指标...\n")
# 自动检测线粒体基因模式
mt_patterns <- c("^MT-", "^mt-", "^MT\\.", "^mt\\.", "MT:", "mt:", "^MT_", "^mt_")
mt_found <- FALSE
for (pattern in mt_patterns) {
mt_genes <- grep(pattern, rownames(seurat_obj), value = TRUE)
if (length(mt_genes) > 0) {
cat(paste(" 检测到线粒体基因模式:", pattern, "(", length(mt_genes), "个基因)\n"))
seurat_obj[["percent.mt"]] <- PercentageFeatureSet(seurat_obj, pattern = pattern)
mt_found <- TRUE
break
}
}
if (!mt_found) {
cat(" 警告: 未检测到线粒体基因,跳过线粒体百分比计算\n")
seurat_obj[["percent.mt"]] <- 0
}
# 自动检测核糖体基因模式
rb_patterns <- c("^RP[SL]", "^Rp[sl]", "^RPS", "^RPL", "^rps", "^rpl")
rb_found <- FALSE
for (pattern in rb_patterns) {
rb_genes <- grep(pattern, rownames(seurat_obj), value = TRUE)
if (length(rb_genes) > 0) {
cat(paste(" 检测到核糖体基因模式:", pattern, "(", length(rb_genes), "个基因)\n"))
seurat_obj[["percent.rb"]] <- PercentageFeatureSet(seurat_obj, pattern = pattern)
rb_found <- TRUE
break
}
}
if (!rb_found) {
cat(" 警告: 未检测到核糖体基因,跳过核糖体百分比计算\n")
seurat_obj[["percent.rb"]] <- 0
}
# 5. 可视化质控指标
cat("\n5. 生成质控可视化...\n")
# 创建质控图
qc_plots <- list()
# 样本细胞数分布(按分组着色)
qc_plots$sample_bar <- ggplot(seurat_obj@meta.data,
aes(x = sample, fill = group)) +
geom_bar() +
theme_bw() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(title = "cell number per sample", x = "sample", y = "cell number", fill = "group")
# 质控指标小提琴图
qc_features <- c("nFeature_RNA", "nCount_RNA")
if (mt_found) qc_features <- c(qc_features, "percent.mt")
if (rb_found) qc_features <- c(qc_features, "percent.rb")
# 自定义函数创建小提琴图
create_custom_vlnplot <- function(seurat_obj, features, group.by = "sample", pt.size = 0.1) {
# 提取数据
plot_data <- FetchData(seurat_obj, vars = c(features, group.by))
# 转换为长格式
library(reshape2)
plot_data_long <- melt(plot_data, id.vars = group.by)
colnames(plot_data_long) <- c("group", "feature", "value")
# 创建小提琴图
p <- ggplot(plot_data_long, aes(x = group, y = value, fill = group)) +
geom_violin(scale = "width", trim = TRUE) +
geom_boxplot(width = 0.1, fill = "white", outlier.shape = NA) +
facet_wrap(~feature, scales = "free_y", ncol = min(4, length(features))) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none",
strip.background = element_rect(fill = "lightgray")
) +
labs(x = "", y = "Value")
return(p)
}
qc_plots$vln_features <- create_custom_vlnplot(
seurat_obj,
features = qc_features,
group.by = "sample"
)
# 保存质控图
pdf(file.path(args$output, "2_qc_plots.pdf"), width = 14, height = 10)
print(qc_plots$sample_bar)
print(qc_plots$vln_features)
dev.off()
# 6. 细胞过滤
cat("\n6. 过滤低质量细胞...\n")
cat(paste("过滤前细胞数:", ncol(seurat_obj), "\n"))
# 创建过滤条件
keep_cells <- seurat_obj$nFeature_RNA >= args$min_features &
seurat_obj$nFeature_RNA <= args$max_features
if (mt_found) {
keep_cells <- keep_cells & seurat_obj$percent.mt <= args$max_mt
}
if (rb_found) {
keep_cells <- keep_cells & seurat_obj$percent.rb <= args$max_rb
}
seurat_obj <- seurat_obj[,names(keep_cells)]
cat(paste("过滤后细胞数:", ncol(seurat_obj), "\n"))
cat(paste("移除细胞数:", sum(!keep_cells), "\n"))
# 保存过滤后数据
saveRDS(seurat_obj, file.path(args$output, "3_filtered.rds"))
cat(paste("过滤后数据已保存到:", file.path(args$output, "3_filtered.rds"), "\n"))
# 7. 标准化和特征选择
cat("\n7. 数据标准化和特征选择...\n")
# 标准化
seurat_obj <- NormalizeData(
seurat_obj,
normalization.method = "LogNormalize",
scale.factor = 10000,
verbose = FALSE
)
# 寻找高变基因
seurat_obj <- FindVariableFeatures(
seurat_obj,
selection.method = "vst",
nfeatures = args$nfeatures,
verbose = FALSE
)
# 8. 数据缩放和PCA
cat("\n8. 数据缩放和PCA降维...\n")
# 数据缩放
seurat_obj <- ScaleData(
seurat_obj,
verbose = FALSE
)
# PCA降维
seurat_obj <- RunPCA(
seurat_obj,
features = VariableFeatures(object = seurat_obj),
npcs = args$npcs,
verbose = FALSE
)
# PCA可视化
pca_plots <- list()
pca_plots$elbow <- ElbowPlot(seurat_obj, ndims = args$npcs)
pca_plots$pca_by_sample <- DimPlot(
seurat_obj,
reduction = "pca",
group.by = "sample",
dims = c(1, 2)
) + ggtitle("PCA - color by sample")
pca_plots$pca_by_group <- DimPlot(
seurat_obj,
reduction = "pca",
group.by = "group",
dims = c(1, 2)
) + ggtitle("PCA - color by sample")
pdf(file.path(args$output, "4_pca_analysis.pdf"), width = 14, height = 10)
print(pca_plots$elbow)
print(pca_plots$pca_by_sample)
print(pca_plots$pca_by_group)
dev.off()
# 9. Harmony批次校正
cat("\n9. Harmony批次校正...\n")
# 使用Seurat V5的IntegrateLayers进行Harmony整合
seurat_obj <- IntegrateLayers(
object = seurat_obj,
method = HarmonyIntegration,
orig.reduction = "pca",
new.reduction = "harmony",
verbose = FALSE
)
cat(" Harmony整合完成!\n")
# 保存Harmony结果
saveRDS(seurat_obj, file.path(args$output, "5_harmony_integrated.rds"))
cat(paste("Harmony整合数据已保存到:", file.path(args$output, "5_harmony_integrated.rds"), "\n"))
# 10. UMAP降维
cat("\n10. UMAP降维...\n")
# 基于Harmony的UMAP
seurat_obj <- RunUMAP(
seurat_obj,
reduction = "harmony",
dims = 1:args$ndims,
reduction.name = "umap_harmony",
reduction.key = "UMAPHARMONY_",
verbose = FALSE
)
# 11. 聚类分析
cat("\n11. 细胞聚类...\n")
# 基于Harmony的聚类
seurat_obj <- FindNeighbors(
seurat_obj,
reduction = "harmony",
dims = 1:args$ndims,
graph.name = "harmony_snn",
verbose = FALSE
)
seurat_obj <- FindClusters(
seurat_obj,
resolution = args$resolution,
graph.name = "harmony_snn",
algorithm = 1,
verbose = FALSE
)
# 重命名聚类列
seurat_obj$seurat_clusters <- seurat_obj$seurat_clusters
# 12. 结果可视化
cat("\n12. 生成结果可视化...\n")
# 创建可视化图集
viz_plots <- list()
# UMAP可视化
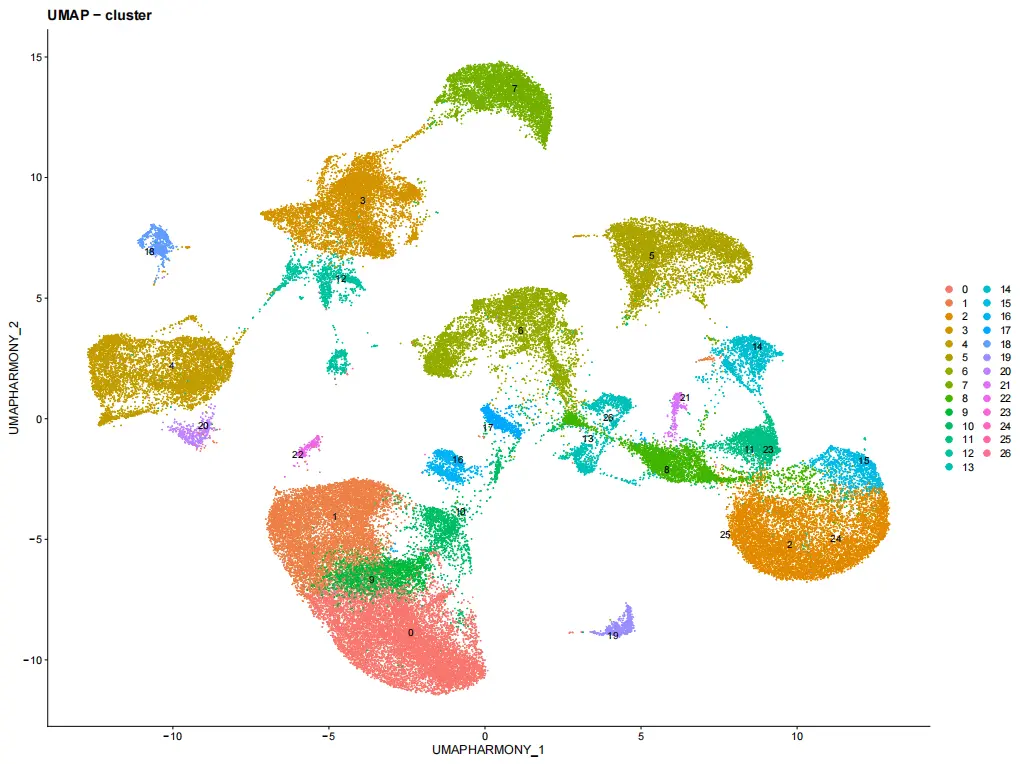
viz_plots$umap_clusters <- DimPlot(
seurat_obj,
reduction = "umap_harmony",
label = TRUE,
repel = TRUE,
label.size = 4
) + ggtitle("UMAP - cluster")
viz_plots$umap_samples <- DimPlot(
seurat_obj,
reduction = "umap_harmony",
group.by = "sample",
shuffle = TRUE
) + ggtitle("UMAP - sample")
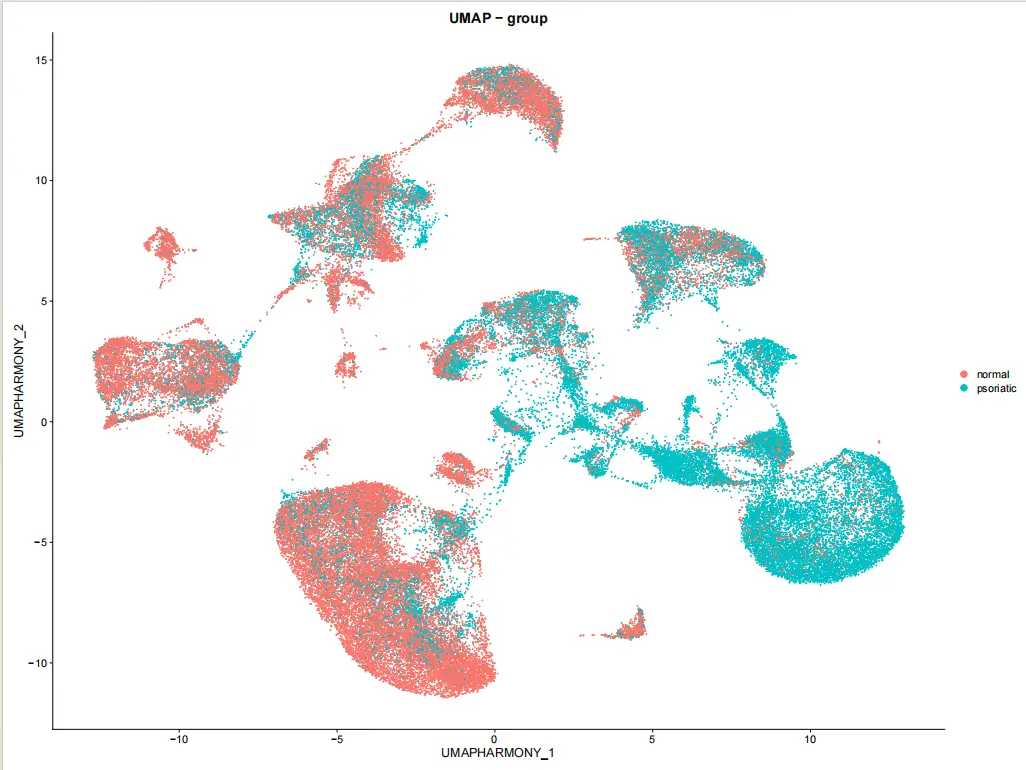
viz_plots$umap_groups <- DimPlot(
seurat_obj,
reduction = "umap_harmony",
group.by = "group",
shuffle = TRUE
) + ggtitle("UMAP - group")
# 样本在聚类中的分布
viz_plots$cluster_by_sample <- ggplot(
seurat_obj@meta.data,
aes(x = seurat_clusters, fill = sample)
) +
geom_bar(position = "fill") +
theme_bw() +
labs(x = "Cluster", y = "Proportion", title = "Proportion for sample") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# 分组在聚类中的分布
viz_plots$cluster_by_group <- ggplot(
seurat_obj@meta.data,
aes(x = seurat_clusters, fill = group)
) +
geom_bar(position = "fill") +
theme_bw() +
labs(x = "Cluster", y = "Proportion", title = "Proportion for group") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# 保存可视化
pdf(file.path(args$output, "6_harmony_results.pdf"), width = 16, height = 12)
print(viz_plots$umap_clusters)
print(viz_plots$umap_samples)
print(viz_plots$umap_groups)
print(viz_plots$cluster_by_sample)
print(viz_plots$cluster_by_group)
dev.off()
# 13. 保存最终结果
cat("\n13. 保存最终结果...\n")
# 保存最终RDS文件
saveRDS(seurat_obj, file.path(args$output, "7_final_seurat_object.rds"))
cat(paste("最终数据已保存到:", file.path(args$output, "7_final_seurat_object.rds"), "\n"))
# 保存元数据
metadata <- seurat_obj@meta.data
write.csv(metadata, file.path(args$output, "8_cell_metadata.csv"), row.names = TRUE)
cat(paste("细胞元数据已保存到:", file.path(args$output, "8_cell_metadata.csv"), "\n"))
# 14. 寻找marker基因
cat("\n14. 寻找marker基因...\n")
# 设置默认assay为RNA
DefaultAssay(seurat_obj) <- "RNA"
seurat_obj <- NormalizeData(seurat_obj, verbose = FALSE)
# 寻找所有cluster的marker基因
markers <- FindAllMarkers(
seurat_obj,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25,
verbose = FALSE
)
# 保存marker基因
write.csv(markers, file.path(args$output, "9_cluster_markers.csv"))
cat(paste("聚类marker基因已保存到:", file.path(args$output, "9_cluster_markers.csv"), "\n"))
# 15. 生成分析报告
cat("\n15. 生成分析报告...\n")
sink(file.path(args$output, "10_analysis_report.txt"))
cat("Seurat V5 + Harmony 批次校正分析报告\n")
cat("=====================================\n")
cat(paste("分析时间:", Sys.time(), "\n"))
cat(paste("样本数量:", length(seurat_list), "\n"))
cat(paste("数据格式统计:\n"))
cat(paste(" 10X格式:", sum(samples_df$data_type == "10X"), "\n"))
cat(paste(" CSV格式:", sum(samples_df$data_type == "csv"), "\n"))
cat(paste("分组统计:\n"))
print(table(samples_df$group))
cat(paste("\n细胞数量统计:\n"))
cat(paste(" 合并后细胞数:", ncol(seurat_obj) + sum(!keep_cells), "\n"))
cat(paste(" 过滤后细胞数:", ncol(seurat_obj), "\n"))
cat(paste(" 移除细胞数:", sum(!keep_cells), "\n"))
cat(paste("\n聚类结果:\n"))
cat(paste(" 聚类数:", length(unique(seurat_obj$seurat_clusters)), "\n"))
cat(paste(" 聚类分辨率:", args$resolution, "\n"))
cat("\n各聚类细胞数:\n")
print(table(seurat_obj$seurat_clusters))
cat("\n数据类型检测:\n")
cat(paste(" 线粒体基因检测:", mt_found, "\n"))
cat(paste(" 核糖体基因检测:", rb_found, "\n"))
cat("\n输出文件清单:\n")
output_files <- list.files(args$output, pattern = "\\.(rds|csv|pdf|txt)$")
for (file in output_files) {
file_info <- file.info(file.path(args$output, file))
cat(paste(" ", file, " (", round(file_info$size/1024/1024, 2), " MB)\n"))
}
sink()
cat("\n========================================\n")
cat("分析完成!所有结果已保存到:", args$output, "\n")
cat("========================================\n")
#!/usr/bin/env python
# Scanpy + Harmony 批次校正流程 - 支持CSV和10X格式
# 使用: python scanpy_harmony.py --samples sample.txt --output results
import argparse
import os
import sys
import warnings
import numpy as np
import pandas as pd
import scanpy as sc
import matplotlib.pyplot as plt
import seaborn as sns
from scipy import sparse
import anndata
import logging
from typing import List, Dict, Optional
# 设置日志
logging.basicConfig(level=logging.INFO, format='%(asctime)s - %(levelname)s - %(message)s')
logger = logging.getLogger(__name__)
# 忽略警告
warnings.filterwarnings('ignore')
def parse_arguments():
"""解析命令行参数"""
parser = argparse.ArgumentParser(description='Scanpy + Harmony 批次校正流程 - 支持CSV和10X格式')
parser.add_argument('--samples', type=str, required=True,
help='样本信息文件,四列:样本名、数据路径、数据类型(10X/csv)、分组信息')
parser.add_argument('--output', type=str, default='harmony_results',
help='输出目录路径')
parser.add_argument('--min_cells', type=int, default=3,
help='基因至少在多少细胞中表达')
parser.add_argument('--min_genes', type=int, default=200,
help='细胞至少检测到多少基因')
parser.add_argument('--max_genes', type=int, default=5000,
help='细胞最多检测到多少基因')
parser.add_argument('--max_mt', type=float, default=20,
help='最大线粒体基因百分比')
parser.add_argument('--max_rb', type=float, default=50,
help='最大核糖体基因百分比')
parser.add_argument('--n_top_genes', type=int, default=2000,
help='高变基因数量')
parser.add_argument('--n_pcs', type=int, default=50,
help='PCA计算维度')
parser.add_argument('--n_neighbors', type=int, default=15,
help='最近邻数量')
parser.add_argument('--resolution', type=float, default=0.8,
help='聚类分辨率')
parser.add_argument('--harmony_key', type=str, default='sample',
help='Harmony校正的关键变量')
parser.add_argument('--harmony_max_iter', type=int, default=20,
help='Harmony最大迭代次数')
parser.add_argument('--threads', type=int, default=4,
help='并行线程数')
parser.add_argument('--csv_sep', type=str, default=',',
choices=[',', ';', '\t', ' '],
help='CSV文件分隔符')
parser.add_argument('--csv_has_index', action='store_true',
help='CSV文件是否有索引(基因名)')
parser.add_argument('--csv_has_header', action='store_true',
help='CSV文件是否有表头(细胞名)')
return parser.parse_args()
def read_sc_data(data_path: str, data_type: str, sample_name: str,
sep: str = ',', has_index: bool = True, has_header: bool = True) -> anndata.AnnData:
"""根据数据类型读取数据"""
logger.info(f" 读取 {data_type} 格式数据: {data_path}")
if data_type == "10X":
# 10X Genomics格式
if not os.path.isdir(data_path):
raise FileNotFoundError(f"10X数据目录不存在: {data_path}")
# 读取10X数据
try:
adata = sc.read_10x_mtx(data_path, var_names='gene_symbols', cache=True)
logger.info(f" 成功读取10X数据,矩阵维度: {adata.shape[0]} × {adata.shape[1]}")
except Exception as e:
# 尝试其他方法
logger.warning(f" sc.read_10x_mtx失败,尝试其他方法: {e}")
try:
# 尝试读取matrix.mtx文件
matrix_file = os.path.join(data_path, 'matrix.mtx.gz')
if os.path.exists(matrix_file):
adata = sc.read_mtx(matrix_file)
logger.info(f" 成功读取.mtx.gz格式")
else:
raise FileNotFoundError(f"未找到matrix.mtx.gz文件")
except Exception as e2:
raise RuntimeError(f"无法读取10X数据: {data_path}\n错误信息: {e2}")
elif data_type == "csv":
# CSV格式
if not os.path.isfile(data_path):
raise FileNotFoundError(f"CSV文件不存在: {data_path}")
logger.info(f" 读取CSV文件,分隔符: {sep}")
# 读取CSV文件
try:
df = pd.read_csv(data_path, sep=sep,
index_col=0 if has_index else None,
header=0 if has_header else None)
# 转换为AnnData对象
# 注意:Scanpy期望行为细胞,列为基因
# 如果CSV是基因×细胞,需要转置
if df.shape[0] > df.shape[1]: # 通常基因数 > 细胞数
logger.info(" 检测到基因×细胞格式,进行转置...")
df = df.T
# 创建AnnData对象
adata = anndata.AnnData(X=df.values,
obs=pd.DataFrame(index=df.index),
var=pd.DataFrame(index=df.columns))
logger.info(f" 成功读取CSV数据,矩阵维度: {adata.shape[0]} × {adata.shape[1]}")
except Exception as e:
raise RuntimeError(f"读取CSV文件失败: {data_path}\n错误信息: {e}")
else:
raise ValueError(f"不支持的数据类型: {data_type}。支持的类型: 10X, csv")
return adata
def calculate_qc_metrics(adata: anndata.AnnData) -> anndata.AnnData:
"""计算质量控制指标"""
# 计算基本QC指标
sc.pp.calculate_qc_metrics(adata, percent_top=None, log1p=False, inplace=True)
# 检测线粒体基因
mt_patterns = ['^MT-', '^mt-', '^MT\.', '^mt\.', 'MT:', 'mt:', '^MT_', '^mt_']
mt_genes_found = False
for pattern in mt_patterns:
mt_genes = adata.var_names.str.contains(pattern)
if mt_genes.any():
logger.info(f" 检测到线粒体基因模式: {pattern} ({mt_genes.sum()}个基因)")
# 计算线粒体基因百分比
if sparse.issparse(adata.X):
mt_counts = adata[:, mt_genes].X.sum(axis=1).A1
total_counts = adata.X.sum(axis=1).A1
else:
mt_counts = adata[:, mt_genes].X.sum(axis=1)
total_counts = adata.X.sum(axis=1)
adata.obs['pct_counts_mt'] = mt_counts / total_counts * 100
mt_genes_found = True
break
if not mt_genes_found:
logger.warning(" 未检测到线粒体基因,跳过线粒体百分比计算")
adata.obs['pct_counts_mt'] = 0
# 检测核糖体基因
rb_patterns = ['^RP[SL]', '^Rp[sl]', '^RPS', '^RPL', '^rps', '^rpl']
rb_genes_found = False
for pattern in rb_patterns:
rb_genes = adata.var_names.str.contains(pattern)
if rb_genes.any():
logger.info(f" 检测到核糖体基因模式: {pattern} ({rb_genes.sum()}个基因)")
# 计算核糖体基因百分比
if sparse.issparse(adata.X):
rb_counts = adata[:, rb_genes].X.sum(axis=1).A1
total_counts = adata.X.sum(axis=1).A1
else:
rb_counts = adata[:, rb_genes].X.sum(axis=1)
total_counts = adata.X.sum(axis=1)
adata.obs['pct_counts_rb'] = rb_counts / total_counts * 100
rb_genes_found = True
break
if not rb_genes_found:
logger.warning(" 未检测到核糖体基因,跳过核糖体百分比计算")
adata.obs['pct_counts_rb'] = 0
return adata
def main():
"""主函数"""
# 解析参数
args = parse_arguments()
# 创建输出目录
os.makedirs(args.output, exist_ok=True)
# 设置Scanpy参数
sc.settings.verbosity = 3 # 详细信息
sc.settings.set_figure_params(dpi=300, facecolor='white',
figsize=(10, 8), fontsize=12)
sc.settings.figdir = args.output
# 设置并行
if args.threads > 1:
sc._settings.ScanpyConfig.n_jobs = args.threads
# 记录参数
logger.info("="*50)
logger.info("Scanpy + Harmony 批次校正流程参数设置:")
logger.info("="*50)
logger.info(f"样本文件: {args.samples}")
logger.info(f"输出目录: {args.output}")
logger.info(f"数据格式支持: 10X, csv")
logger.info(f"CSV分隔符: {args.csv_sep}")
logger.info(f"CSV有索引: {args.csv_has_index}")
logger.info(f"CSV有表头: {args.csv_has_header}")
logger.info(f"Harmony校正变量: {args.harmony_key}")
logger.info(f"最小细胞数: {args.min_cells}")
logger.info(f"最小基因数: {args.min_genes}")
logger.info(f"最大基因数: {args.max_genes}")
logger.info(f"最大线粒体百分比: {args.max_mt}%")
logger.info(f"最大核糖体百分比: {args.max_rb}%")
logger.info(f"高变基因数: {args.n_top_genes}")
logger.info(f"PCA维度: {args.n_pcs}")
logger.info(f"最近邻数量: {args.n_neighbors}")
logger.info(f"聚类分辨率: {args.resolution}")
logger.info(f"Harmony最大迭代: {args.harmony_max_iter}")
logger.info(f"并行线程数: {args.threads}")
logger.info("="*50)
# 1. 读取样本信息
logger.info("1. 读取样本信息...")
samples_df = pd.read_csv(args.samples, sep='\t', header=None,
names=['sample_name', 'data_path', 'data_type', 'group'])
# 验证数据类型
valid_types = ['10X', 'csv']
invalid_types = set(samples_df['data_type']) - set(valid_types)
if invalid_types:
raise ValueError(f"无效的数据类型: {invalid_types}。有效类型: {valid_types}")
logger.info(f"找到 {len(samples_df)} 个样本:")
for _, row in samples_df.iterrows():
logger.info(f" {row['sample_name']}: {row['data_path']} [{row['data_type']}] 分组: {row['group']}")
# 2. 加载和合并数据
logger.info("\n2. 加载单细胞数据...")
adata_list = []
for _, row in samples_df.iterrows():
sample_name = row['sample_name']
data_path = row['data_path']
data_type = row['data_type']
group_info = row['group']
logger.info(f" 加载样本: {sample_name} [{data_type}] 分组: {group_info}")
# 读取数据
adata = read_sc_data(
data_path=data_path,
data_type=data_type,
sample_name=sample_name,
sep=args.csv_sep,
has_index=args.csv_has_index,
has_header=args.csv_has_header
)
# 添加样本信息和分组信息
adata.obs['sample'] = sample_name
adata.obs['group'] = group_info
adata.obs['orig_ident'] = sample_name
# 确保细胞名唯一
adata.obs_names = [f"{sample_name}_{cell_id}" for cell_id in adata.obs_names]
adata_list.append(adata)
logger.info(f" 细胞数: {adata.shape[0]}, 基因数: {adata.shape[1]}")
# 清理内存
del adata
# 3. 合并所有样本
logger.info("\n3. 合并所有样本...")
if len(adata_list) == 1:
adata = adata_list[0]
else:
# 合并AnnData对象
adata = adata_list[0].concatenate(
adata_list[1:],
join='inner',
batch_key='sample',
batch_categories=samples_df['sample_name'].tolist()
)
logger.info(f"合并后总细胞数: {adata.shape[0]}")
logger.info(f"合并后总基因数: {adata.shape[1]}")
logger.info(f"数据来源统计:")
logger.info(f" 10X格式样本: {(samples_df['data_type'] == '10X').sum()}")
logger.info(f" CSV格式样本: {(samples_df['data_type'] == 'csv').sum()}")
logger.info(f"分组信息统计:")
group_counts = samples_df['group'].value_counts()
for group_name, count in group_counts.items():
logger.info(f" {group_name}: {count}个样本")
# 保存原始数据
adata.write(os.path.join(args.output, '1_merged_raw.h5ad'))
logger.info(f"原始数据已保存到: {os.path.join(args.output, '1_merged_raw.h5ad')}")
# 4. 计算质量控制指标
logger.info("\n4. 计算质量控制指标...")
adata = calculate_qc_metrics(adata)
# 5. 可视化质控指标
logger.info("\n5. 生成质控可视化...")
# 使用sc.pl创建质控图
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'pct_counts_rb'],
groupby='sample', rotation=45, log=True, stripplot=False,
save='_qc_violin.png')
# 样本细胞数分布
plt.figure(figsize=(12, 6))
sample_counts = adata.obs['sample'].value_counts()
plt.bar(sample_counts.index, sample_counts.values)
plt.title('Cell Number per Sample')
plt.xlabel('Sample')
plt.ylabel('Cell Count')
plt.xticks(rotation=45)
plt.tight_layout()
plt.savefig(os.path.join(args.output, 'sample_counts.png'), dpi=300)
plt.close()
# 6. 细胞过滤
logger.info("\n6. 过滤低质量细胞...")
logger.info(f"过滤前细胞数: {adata.shape[0]}")
# 创建过滤条件
if 'n_genes_by_counts' in adata.obs.columns:
min_genes_mask = adata.obs['n_genes_by_counts'] >= args.min_genes
max_genes_mask = adata.obs['n_genes_by_counts'] <= args.max_genes
else:
# 如果没有计算基因数,计算一下
n_genes = (adata.X > 0).sum(axis=1)
n_genes = np.array(n_genes).flatten()
adata.obs['n_genes_by_counts'] = n_genes
min_genes_mask = n_genes >= args.min_genes
max_genes_mask = n_genes <= args.max_genes
if 'pct_counts_mt' in adata.obs.columns:
mt_mask = adata.obs['pct_counts_mt'] <= args.max_mt
else:
mt_mask = pd.Series(True, index=adata.obs.index)
if 'pct_counts_rb' in adata.obs.columns:
rb_mask = adata.obs['pct_counts_rb'] <= args.max_rb
else:
rb_mask = pd.Series(True, index=adata.obs.index)
# 合并所有条件
keep_cells = min_genes_mask & max_genes_mask & mt_mask & rb_mask
# 应用过滤
adata = adata[keep_cells, :].copy()
logger.info(f"过滤后细胞数: {adata.shape[0]}")
logger.info(f"移除细胞数: {sum(~keep_cells)}")
# 保存过滤后数据
adata.write(os.path.join(args.output, '2_filtered.h5ad'))
logger.info(f"过滤后数据已保存到: {os.path.join(args.output, '2_filtered.h5ad')}")
# 7. 标准化和特征选择
logger.info("\n7. 数据标准化和特征选择...")
# 标准化
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 寻找高变基因
sc.pp.highly_variable_genes(adata, n_top_genes=args.n_top_genes, flavor='seurat')
# 8. 数据缩放和PCA
logger.info("\n8. 数据缩放和PCA降维...")
# 数据缩放(处理稀疏矩阵警告)
logger.info("进行数据缩放...")
if sparse.issparse(adata.X):
logger.info("检测到稀疏矩阵,转换为密集矩阵以提高性能...")
adata.X = adata.X.toarray()
sc.pp.scale(adata, max_value=10)
# PCA降维
logger.info(f"进行PCA降维,维度: {args.n_pcs}")
sc.pp.pca(adata, n_comps=args.n_pcs, use_highly_variable=True)
# PCA可视化
logger.info("生成PCA可视化...")
# 1. 肘部图
plt.figure(figsize=(8, 6))
pca_variance = adata.uns['pca']['variance_ratio']
plt.plot(range(1, len(pca_variance) + 1), pca_variance, 'bo-')
plt.xlabel('Principal Components')
plt.ylabel('Variance Ratio')
plt.title('PCA Variance Ratio')
plt.grid(True, alpha=0.3)
plt.tight_layout()
plt.savefig(os.path.join(args.output, 'pca_variance_ratio.png'), dpi=300)
plt.close()
# 2. PCA散点图(按样本)
sc.pl.pca(adata, color='sample', save='_by_sample.png')
# 3. PCA散点图(按分组)
sc.pl.pca(adata, color='group', save='_by_group.png')
# 9. Harmony批次校正
logger.info("\n9. Harmony批次校正...")
# 检查是否安装了harmony
try:
import scanpy.external as sce
# 使用sc.external.pp.harmony_integrate进行Harmony整合
logger.info("使用sc.external.pp.harmony_integrate进行批次校正...")
# 运行Harmony整合
sce.pp.harmony_integrate(
adata,
key=args.harmony_key,
basis='X_pca',
max_iter_harmony=args.harmony_max_iter,
verbose=True
)
# Harmony结果保存在 adata.obsm['X_pca_harmony']
logger.info("Harmony整合完成!PCA校正后的结果保存在 adata.obsm['X_pca_harmony']")
except ImportError as e:
logger.error(f"导入harmony模块失败: {e}")
logger.error("请安装harmony: pip install harmony-pytorch")
logger.error("或使用scanpy的外部工具: pip install scanpy[harmony]")
sys.exit(1)
# 保存Harmony结果
adata.write(os.path.join(args.output, '3_harmony_integrated.h5ad'))
logger.info(f"Harmony整合数据已保存到: {os.path.join(args.output, '3_harmony_integrated.h5ad')}")
# 10. 邻居图和UMAP降维
logger.info("\n10. UMAP降维...")
# 基于Harmony的邻居图
if 'X_pca_harmony' in adata.obsm:
logger.info("使用Harmony校正后的PCA进行邻居图计算...")
sc.pp.neighbors(adata, n_neighbors=args.n_neighbors, n_pcs=args.n_pcs,
use_rep='X_pca_harmony')
else:
logger.warning("未找到X_pca_harmony,使用原始PCA")
sc.pp.neighbors(adata, n_neighbors=args.n_neighbors, n_pcs=args.n_pcs)
# UMAP降维
sc.tl.umap(adata)
# 11. 聚类分析
logger.info("\n11. 细胞聚类...")
# Leiden聚类
sc.tl.leiden(adata, resolution=args.resolution, key_added='leiden')
# 12. 结果可视化
logger.info("\n12. 生成结果可视化...")
# 使用sc.pl创建UMAP图
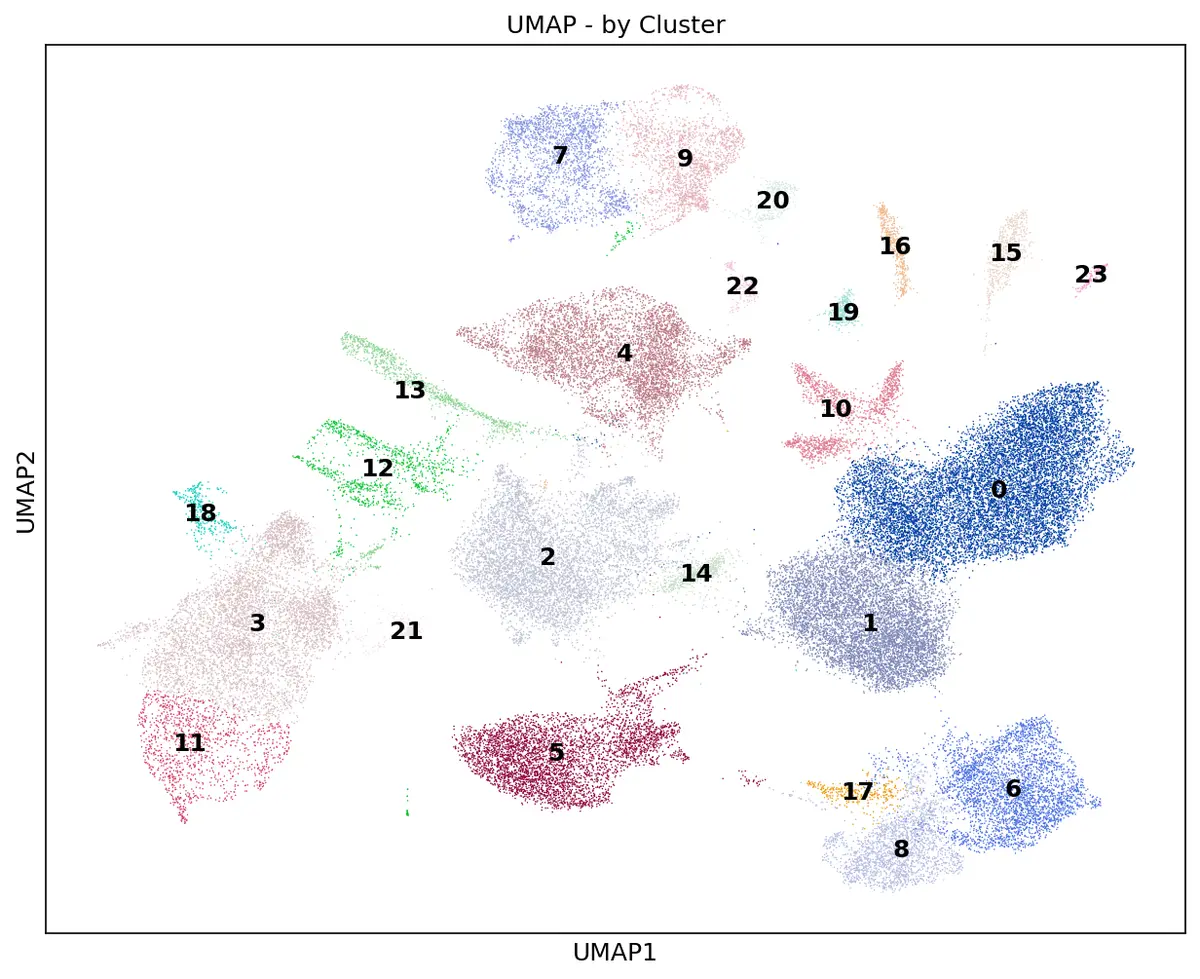
# 1. UMAP按聚类着色
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='UMAP - by Cluster',
save='_umap_clusters.png')
# 2. UMAP按样本着色
sc.pl.umap(adata, color='sample', title='UMAP - by Sample',
save='_umap_samples.png')
# 3. UMAP按分组着色
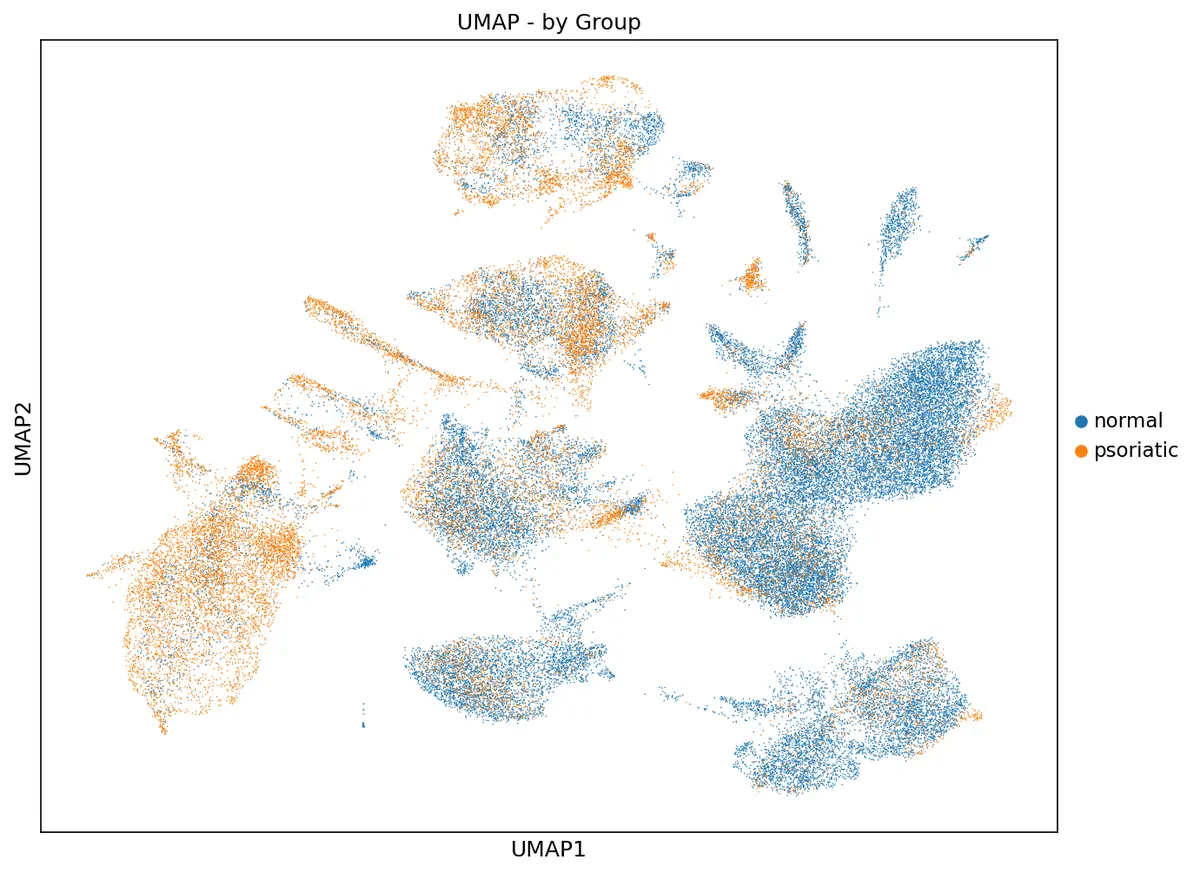
sc.pl.umap(adata, color='group', title='UMAP - by Group',
save='_umap_groups.png')
# 13. 保存最终结果
logger.info("\n13. 保存最终结果...")
# 保存最终h5ad文件
adata.write(os.path.join(args.output, '4_final_adata.h5ad'))
logger.info(f"最终数据已保存到: {os.path.join(args.output, '4_final_adata.h5ad')}")
# 保存元数据
metadata = adata.obs.copy()
metadata.to_csv(os.path.join(args.output, '5_cell_metadata.csv'))
logger.info(f"细胞元数据已保存到: {os.path.join(args.output, '5_cell_metadata.csv')}")
# 14. 寻找marker基因
logger.info("\n14. 寻找marker基因...")
# 设置原始counts用于差异表达分析
adata_raw = adata.copy()
sc.pp.normalize_total(adata_raw, target_sum=1e4)
sc.pp.log1p(adata_raw)
# 寻找所有cluster的marker基因
logger.info("寻找差异表达基因...")
sc.tl.rank_genes_groups(adata_raw, 'leiden', method='wilcoxon', use_raw=False)
# 保存marker基因
# 提取marker基因结果
result = adata_raw.uns['rank_genes_groups']
groups = result['names'].dtype.names
# 创建DataFrame保存所有marker基因
markers_df = pd.DataFrame()
for group in groups:
# 获取该cluster的top genes
names = result['names'][group][:100] # 取前100个
scores = result['scores'][group][:100]
pvals = result['pvals'][group][:100]
pvals_adj = result['pvals_adj'][group][:100]
logfc = result['logfoldchanges'][group][:100]
# 创建临时DataFrame
temp_df = pd.DataFrame({
'cluster': group,
'gene': names,
'score': scores,
'pval': pvals,
'pval_adj': pvals_adj,
'logfc': logfc
})
markers_df = pd.concat([markers_df, temp_df], ignore_index=True)
# 保存到CSV
markers_df.to_csv(os.path.join(args.output, '6_cluster_markers.csv'), index=False)
logger.info(f"聚类marker基因已保存到: {os.path.join(args.output, '6_cluster_markers.csv')}")
# 可视化top marker基因
logger.info("生成marker基因热图...")
sc.pl.rank_genes_groups_heatmap(adata_raw, n_genes=10, groupby='leiden',
show=False, save='_top_markers_heatmap.png',
vmin=-3, vmax=3, cmap='bwr')
# 15. 生成分析报告
logger.info("\n15. 生成分析报告...")
report_file = os.path.join(args.output, '7_analysis_report.txt')
with open(report_file, 'w') as f:
f.write("Scanpy + Harmony 批次校正分析报告\n")
f.write("="*50 + "\n")
f.write(f"分析时间: {pd.Timestamp.now()}\n")
f.write(f"样本数量: {len(adata_list)}\n")
f.write(f"数据格式统计:\n")
f.write(f" 10X格式: {(samples_df['data_type'] == '10X').sum()}\n")
f.write(f" CSV格式: {(samples_df['data_type'] == 'csv').sum()}\n")
f.write(f"分组统计:\n")
f.write(group_counts.to_string() + "\n")
f.write(f"\n细胞数量统计:\n")
f.write(f" 合并后细胞数: {adata.shape[0] + sum(~keep_cells)}\n")
f.write(f" 过滤后细胞数: {adata.shape[0]}\n")
f.write(f" 移除细胞数: {sum(~keep_cells)}\n")
f.write(f"\n聚类结果:\n")
f.write(f" 聚类数: {adata.obs['leiden'].nunique()}\n")
f.write(f" 聚类分辨率: {args.resolution}\n")
f.write(f"\n各聚类细胞数:\n")
f.write(adata.obs['leiden'].value_counts().sort_index().to_string() + "\n")
# 检查质控指标
mt_found = 'pct_counts_mt' in adata.obs.columns and adata.obs['pct_counts_mt'].max() > 0
rb_found = 'pct_counts_rb' in adata.obs.columns and adata.obs['pct_counts_rb'].max() > 0
f.write(f"\n数据类型检测:\n")
f.write(f" 线粒体基因检测: {mt_found}\n")
f.write(f" 核糖体基因检测: {rb_found}\n")
# Harmony整合状态
harmony_success = 'X_pca_harmony' in adata.obsm
f.write(f"\nHarmony整合状态:\n")
f.write(f" 成功整合: {harmony_success}\n")
if harmony_success:
f.write(f" 校正后PCA维度: {adata.obsm['X_pca_harmony'].shape[1]}\n")
# 输出文件清单
f.write(f"\n输出文件清单:\n")
output_files = [f for f in os.listdir(args.output) if f.endswith(('.h5ad', '.csv', '.png', '.txt', '.pdf'))]
for file in sorted(output_files):
file_path = os.path.join(args.output, file)
file_size = os.path.getsize(file_path) / (1024 * 1024) # MB
f.write(f" {file} ({file_size:.2f} MB)\n")
logger.info(f"分析报告已保存到: {report_file}")
logger.info("\n" + "="*50)
logger.info("分析完成!所有结果已保存到: " + args.output)
logger.info("="*50)
if __name__ == "__main__":
main()