在使用 Amber(特别是 AMBER force field + sander/pmemd/ambertools)进行分子动力学(MD)模拟前,蛋白结构的准备 是关键的第一步。Amber 本身不直接提供图形化建模工具,但通过其配套软件包 AmberTools (尤其是 tleap、pdb4amber、antechamber 等)可系统化完成蛋白结构的清洗、补全、质子化、拓扑与坐标文件生成。
本文主要展示和讨论:
- 进行分子动力学模拟前蛋白结构准备的一般流程
- 蛋白结构的准备时加氢原子操作时,使用pdb4amber的reduce还是tleap?
- 蛋白结构中可质子化残基的合理处理
以下是以Amber24操作为例,介绍蛋白(apo,无配体)体系的标准输入文件准备流程。
✅ 一、初始结构准备要求(PDB 文件)
- 来源:实验结构(X-ray,NMR,冷冻电镜Cryo-EM)、同源建模结果或者蛋白结构预测模型(Alphafold,ESMFold等)的结果。
对于来自不同实验类型的结构,参数意义不同。以下是对比:
| 特性 | X-ray晶体学 | Cryo-EM | NMR |
|---|---|---|---|
| 样品环境 | 晶体堆积(非生理) | 玻璃态冰(近生理) | 溶液(生理) |
| 结构数量 | 单一模型 | 单一模型 | 系综(20-100个) |
| 典型分辨率 | 1.0-3.0 Å(<2.0Å为优) | 2.5-4.5 Å(<3.5Å为优) | ~2-5Å(精度指标不同) |
| 动态信息 | 无(B因子间接反映) | 无(局部分辨率反映) | 直接包含在系综中 |
| 大小限制 | 无限制 | <100-200 kDa(传统) | <50-100 kDa |
| 时间尺度 | 静态 | 静态 | ps-ms平均 |
| 温度 | 通常100K | 液氮温度(~100K) | 室温/生理温度 |
- 基本质量要求:
- 完整主链(无缺失残基/原子,尤其 N/Cα/C/O),即研究对象的主链,去处不相关链;
- 残基命名符合标准 IUPAC/Amber 命名(如
ALA,LYS,ASP;非HIS,HID,HIE,HIP需后续正确质子化); - 无重复/冲突的原子序号或链标识;
- 删除不需要或者不关注的链或分子,主要是蛋白结晶附带的与3D结构不相关的配体;
- 建议去除结晶水、配体、辅因子(除非需保留);若需保留,须额外处理。
PDB结构的解读请参考博文:PDB: 结构生物学的"宪法级"格式,但90%使用者从未真正读懂它
蛋白结构预测模型结构的评价指标可参考博文:蛋白结构预测模型评价指标
- ⚠️ 注意事项
- 来自实验结果的PDB结构检查,结构的分辨率 ≤ 2.0 Å(X-ray)、R-factor < 0.20(越低越好);Cryo-EM全局分辨率 ≤ 3.5 Å且活性位点/配体区域局部分辨率高;如果达不到要求,需要注意研究的活性位点或者结合位点相关的残基是否可靠;
- 若为 NMR 结构,选择第一个模型或平均结构;
- 蛋白结构预测模型的PDB结构,可选择置信度高(pLDDT>70,越高越好)的一个或者多个结构。
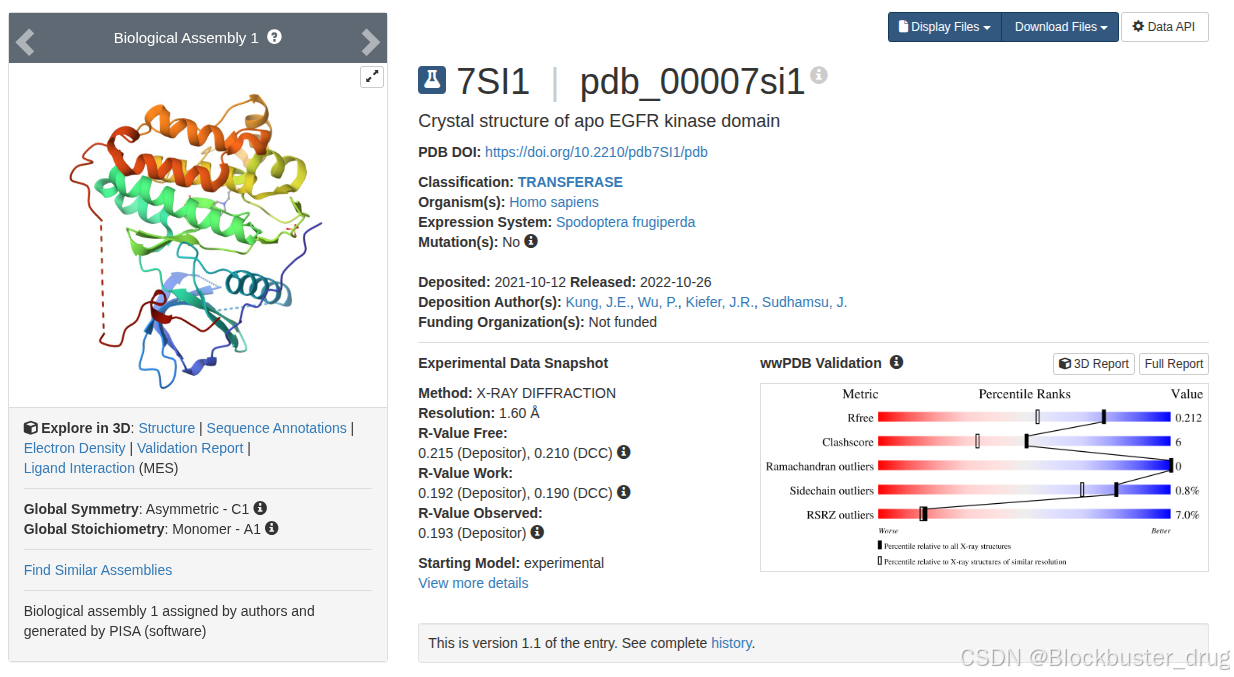
本文以布鲁顿酪氨酸激酶的激酶结构域3D结构为例,PDB ID: 7SI1。
HET MES A1101 12
HET SO4 A1102 5
HET SO4 A1103 5
HETNAM MES 2-(N-MORPHOLINO)-ETHANESULFONIC ACID
HETNAM SO4 SULFATE ION
FORMUL 2 MES C6 H13 N O4 S
FORMUL 3 SO4 2(O4 S 2-)
FORMUL 5 HOH *267(H2 O)该结构中除了蛋白A链331个氨基酸以外,另外有2个硫酸分子,1个MES分子,267个水分子。
A链是我们关注的研究目标,所以去除A链以外的部分。
✅ 二、结构预处理
预处理必不可少。预处理目的:
- 提取关注的结构部分,删除A链以外的分子,只保留蛋白部分的结构;
- 补全实验获得结构中的缺失残基,比如loop等结构,可以使用多个其它程序,比如MOE(structure preparation),Schrodinger(protein preparation),OpenMM中的pdbfixer(基于模板或简单建模的简单修复),
SWISS-MODEL或者Modeller(同源建模)等等; 重命名残基符合Amber读取要求,使用Amber自带的pdb4amber;处理质子化状态:确定可电离残基(His、Asp、Glu、Lys)的质子化状态(pH 7.4为标准);处理 组氨酸(HIS)命名,HID(δ位质子化)、HIE(ε位质子化)、HIP(双质子化)- 对整个蛋白结构的加氢原子的灵魂拷问:使用pdb4amber还是tleap?(讨论见下文)
处理存在的非标准残基,后续将单独介绍,本例不涉及;- 处理二硫键,确认并定义CYS-CYS连接,后续将单独介绍,本例不涉及;
- 处理金属离子,后续将单独介绍,本例不涉及。
本例操作:(不含二硫键,非标准残基和活性位点金属离子或金属离子辅因子)
Step 1: 删除A链以外的分子,只保留蛋白A链结构;使用第三方工具补全缺失的残基,保持蛋白链的完整;删除氢原子,让Amber自动添加。处理过程中去掉了未解析的4个末尾的残基,目前残基总数327;保存结果为7SI1_prep.pdb
Srep 2: 使用pdb4amber,-y 选项即 --nohyd,不保留氢原子,可以留给tleap处理;也可以使用 --reduce选项加氢原子
pdb4amber -i 7SI1_prep.pdb -o 7SI1_clean.pdb --nohydpdb4amber的输出一般包括:
|-----------------|-------------------------|
| pdb4amber输出文件名 | 解释 |
| *.pdb | 残基重新编号的蛋白结构 |
| *_nonprot.pdb | 所有非蛋白组分(配体、离子、结晶水等) |
| *_sslink | 检测到的二硫键信息 |
| *_renum.txt | 残基编号映射记录 |
本例运行后生成4个文件,其中7SI1_clean.pdb,对残基重新编号,从1开始重新编号到327,将作为tleap的输入文件。
没有二硫键和其他非蛋白组分,所以二硫键和非蛋白组分文件为空。
✅ 三、使用 tleap 构建系统(核心步骤)
tleap 是 Amber 的拓扑构建器,负责:
- 加载力场;
- 加载蛋白结构并分配电荷/键级/二面角参数;
- 溶剂化(TIP3P/TIP4P/EW 水模型);
- 中和电荷(添加 Na⁺/Cl⁻ 离子);
- 输出最终输入文件:
prmtop(拓扑)+inpcrd(坐标)。
创建名称为leap.in 脚本,内容如下(#号后面是注释,非必须):
# 加载力场,原子半径参数
source leaprc.protein.ff19SB # 推荐蛋白力场(替代旧 ff14SB)
source leaprc.water.tip3p # 水模型
set default PBradii mbondi2 # 显式设置默认 PBradii 为 mbondi2(推荐)
# 加载蛋白(自动识别残基、加氢、分配质子化态)
mol = loadpdb 7SI1_clean.pdb
# 溶剂化:立方盒,边界距离 10 Å
solvateBox mol TIP3PBOX 10.0
# 中和体系电荷
addions mol Na+ 0
addions mol Cl- 0
# 保存
saveamberparm mol 7SI1_solv.prmtop 7SI1_solv.inpcrd
savepdb mol 7SI1_solv.pdb
# 计算体系电荷
charge mol
quit运行:
tleap -f leap.in运行后输出:
7SI1_solv.prmtop:Amber 拓扑文件(含质量、电荷、键合关系、二面角等);7SI1_solv.inpcrd:初始坐标(含水、离子);7SI1_solv.pdb:可用于可视化验证。
⚠️ 重要注意事项
| 项目 | 说明 |
|---|---|
| 质子化态 | tleap 默认按 pH 7.4 判断(如 ASP→ASH?否,仍为 ASP 去质子化;LYS→LYS 质子化)。His 残基需特别关注------tleap 会尝试智能判断,但建议用 check mol 或 list mol 查看,并结合 pKa 计算(如 propka)人工校验。 |
| 力场选择 | 蛋白首选 ff19SB(增强 backbone χ₁/χ₂ 和 sidechain 构象采样),避免使用过时的 ff99SB。 |
| 水模型匹配 | ff19SB 与 TIP3P 经联合验证,作为优选;若用 TIP4P-EW,需加载 leaprc.water.tip4pew 并确认兼容性。 |
| 周期性边界 | solvateBox 生成正交盒子;如需截角八面体(octahedral),用 solvateOct(节省粒子数)。 |
✅ 四、残基质子化状态与特殊处理
- 在某些情况下,残基质子化状态对MD有重要影响。
- 在 pH 7.4(生理 pH) 条件下,蛋白质中可离子化氨基酸残基的质子化状态主要由其 pKa 值 与环境 pH 的相对关系决定。
- 💡 注意 :AMBER 不会自动判断微环境 pKa !你需要手动指定非标准质子化状态(如将 ASP 改为 ASH 表示质子化)。
- 以下是具有多种质子化状态的氨基酸在AMBER 力场中的默认处理(pH 7.4):
| 残基 | AMBER 默认命名 | 质子化状态 | 说明 |
| Asp | ASP | 去质子化(--COO⁻) | 带 --1 电荷 |
| Glu | GLU | 去质子化(--COO⁻) | 带 --1 电荷 |
| His | HID | Nδ1 质子化 | 最常见默认;也可用 HIE(Nε2 质子化)或 HIP(双质子化) |
| Cys | CYS | 质子化(--SH) | 中性;若形成二硫键 → CYX |
| Tyr | TYR | 质子化(--OH) | 中性 |
| Lys | LYS | 质子化(--NH₃⁺) | 帶 +1 电荷 |
| Arg | ARG | 质子化 | 帶 +1 电荷 |
|---|
- 以下是标准 20 种氨基酸中可离子化残基在 pH 7.4 时的典型质子化状态:
| 残基 | 侧链pKa | pH 7.4时的状态 | Amber残基名 | 电荷 |
|---|---|---|---|---|
| Asp (D) | ~3.9 | 去质子化 (-) | ASP |
-1 |
| Glu (E) | ~4.3 | 去质子化 (-) | GLU |
-1 |
| Lys (K) | ~10.5 | 质子化 (+) | LYS |
+1 |
| Arg (R) | ~12.5 | 质子化 (+) | ARG |
+1 |
| His (H) | ~6.0 | 部分质子化 | HID/HIE/HIP |
0/+1 |
| Cys (C) | ~8.3 | 主要为中性 | CYS |
0 |
| Tyr (Y) | ~10.1 | 质子化 (中性) | TYR |
0 |
| N-terminus | ~8.0 | 质子化 (+) | - | +1 |
| C-terminus | ~3.8 | 去质子化 (-) | - | -1 |
- 但在真实蛋白结构中,局部环境可使 pKa 偏移 ±3 个单位以上:
| 微环境影响 | pKa 变化 | 示例 |
|---|---|---|
| 疏水口袋 | ↑ pKa(更难去质子化) | ASP 在疏水区可能保持质子化(--COOH) |
| 正电荷附近 | ↑ pKa(稳定去质子化形式) | GLU 靠近 ARG → 更易去质子化 |
| 负电荷附近 | ↓ pKa(更难去质子化) | ASP 靠近另一个 ASP → pKa 升高,可能质子化 |
| 金属离子配位 | ↓ pKa(促进去质子化) | CYS-Zn²⁺ → CYS 去质子化(--S⁻) |
| 氢键网络 | 可升可降 | HIS 在氢键供体/受体间,pKa 可调至 7--8 |
- 组氨酸(His)- 特殊情况:
在Amber中,组氨酸由于pKa接近生理状态的pH(7.4),因此有3中不同的质子化状态,单质子化的HIE(为主)和HID,双质子化的HIP:
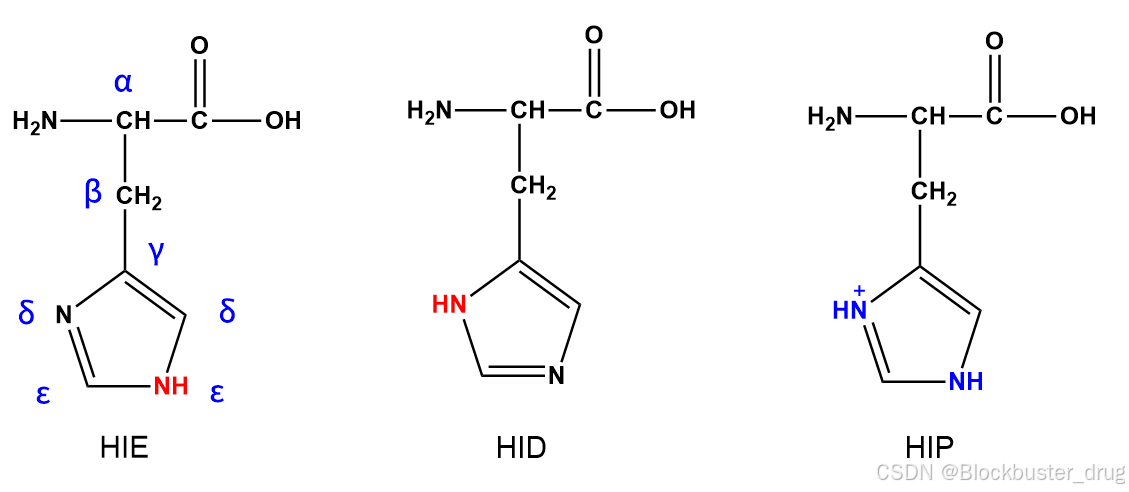
pKa ≈ 6.0,接近生理pH,存在三种状态:
HID(δ位质子化) HIE(ε位质子化) HIP(双质子化)
| | |
ND1-H NE2-H ND1-H + NE2-H
(中性) (中性) (+1电荷)
pH 7.4时的分布:
- ~80-90% 为 HID 或 HIE(单质子化,中性),ε位N碱性强,HIE为主
- ~10-20% 为 HIP(双质子化,带电)
- 具体比例取决于微环境- 半胱氨酸(Cys)- 特殊情况
| 环境 | pH 7.4状态 | Amber处理 |
|---|---|---|
| 游离Cys | 主要为 CYS(-SH,中性) |
标准模板 |
| 二硫键Cys | CYX(氧化态,中性) |
需手动指定或自动检测 |
| 金属配位Cys | 可能去质子化(CYS⁻) | 需自定义参数 |
| 活性位点Cys(如半胱氨酸蛋白酶) | 去质子化(硫醇盐) | 需特殊处理 |
二硫键定义示例:
如果含有二硫键,可以在tleap中使用bond关键字指定,pdb4amber有时不能正确处理。
# tleap中指定二硫键
bond mol.32.SG mol.35.SG # Cys32-Cys35
# 或自动检测(pdb4amber生成sslink文件)-
酪氨酸(Tyr)- 特殊酶活性位点
标准状态:质子化(中性,-OH)
特殊案例:- 酪氨酸激酶活性位点:可能去质子化参与磷酸转移
- 光合作用中心:特殊氢键环境降低pKa
- 需用PROPKA等工具预测异常pKa
-
如何确定你的体系中残基的真实质子化状态?
方法 1:使用 pKa 预测工具(推荐)
PROPKA(集成在 PDB2PQR、ChimeraX 中)
propka3 7SI1_clean.pdb输出名称为7SI1_clean.pka的文件,里面有每个残基的预测 pKa 和建议质子化状态。
方法 2:手动分析(适用于已知机制)
查文献:该蛋白活性位点残基的质子化状态是否已知?
看结构:ASP/GLU 是否在疏水区?HIS 是否与 ASP/Glu 形成氢键?
方法 3:在 AMBER 中测试
分别用不同质子化状态运行短 MD,看哪个更稳定。
- 是否需手动干预?
| 残基 | 溶液中(标准) | 蛋白中(可能) | AMBER 默认 | 是否需手动干预? |
|---|---|---|---|---|
| ASP | 去质子化 (--) | 可能质子化(若疏水) | ASP (--) | ✅ 若在活性位点/疏水区 |
| GLU | 去质子化 (--) | 可能质子化 | GLU (--) | ✅ 同上 |
| HIS | 部分质子化 | HID / HIE / HIP | HID | ✅ 强烈建议检查 |
| CYS | 质子化 (0) | 可能去质子化(若配位金属) | CYS (0) | ✅ 若结合 Zn²⁺/Fe²⁺ |
| LYS | 质子化 (+) | 几乎总是质子化 | LYS (+) | ❌ 通常不用改 |
| ARG | 质子化 (+) | 几乎总是质子化 | ARG (+) | ❌ 通常不用改 |
总之,需要根据研究目的,确定蛋白质必要的质子化状态。重点关注组氨酸、半胱氨酸和酸性氨基酸的质子化状态,是否与研究目标一致。
✅ 五、蛋白结构加氢处理讨论
在 AMBER 工作流中,可以使用pdb4amber的reduce选项调用reduce模块加氢,也可以留给 tleap使用Amber力场参数加氢,是一个常见但关键的选择。两者在加氢策略、质子化状态、适用场景上有本质区别。
| 场景 | 推荐方式 |
|---|---|
| 常规蛋白模拟(无特殊 pH/催化残基) | ✅ 留给 tleap 加氢(更简单、标准) |
| 需要精确控制组氨酸/酸碱残基质子化状态(如酶活性位点) | ✅ 用外部工具(如 reduce / MCPB.py / H++)预加氢,pdb4amber 仅清洗 |
pdb4amber 自身不推荐用于加氢! |
❌ 它没有智能质子化能力 |
两者的本质区别:
| 特性 | pdb4amber --reduce |
tleap 自动加氢 |
|---|---|---|
| 加氢时机 | 预处理阶段(PDB→PDB) | 拓扑生成阶段(PDB→拓扑) |
| 质子化判断 | 基于Reduce算法(环境感知) | 基于残基模板(标准pH 7.0) |
| His处理方式 | 自动判断HID/HIE/HIP | 默认全为HIS(需手动指定) |
| 灵活性 | 高,可人工干预 | 低,依赖模板 |
| 可重复性 | 中等(依赖Reduce版本) | 高(固定模板) |
| 可视化验证 | ✅ 加氢后可检查 | ❌ 直接进模拟 |
两种加氢的详细描述:
1. pdb4amber 预处理加氢
pdb4amber 是AmberTools中专门用于准备PDB文件的工具,它可以在进入tleap前对结构进行"清理"和预处理。pdb4amber 的设计初衷是"清洗 PDB 格式",不是"加氢工具" 。它的 --reduce 选项只是调用外部 reduce 程序,并非内置功能。
特点:
- 修复结构问题:自动修复残基命名、原子命名、缺失重原子等(例如将HIS转换为HID/HIE/HIP)。
- 选择性加氢 :可根据pH值(通过
--reduce选项)添加氢原子,并优化氢原子的取向(尤其是His、Asn、Gln的翻转问题)。 - 去除非标准内容:删除水分子、异质原子(如配体、结晶剂),生成"干净"的蛋白结构。
- 输出标准化PDB:生成符合Amber命名规范的PDB文件(如原子名称、残基顺序)。
典型命令:
pdb4amber -i input.pdb -o cleaned.pdb --reduce # 加氢并优化氢键网络优点:
- 更可控:可先检查修复后的结构,再进入tleap。
- 处理复杂情况:适合晶体结构中存在命名错误、质子化状态不确定的情况。
- 减少tleap报错:避免tleap因原子命名问题而失败。
2. tleap 自动加氢
tleap 是Amber的建模工具,在加载力场和生成拓扑文件时,可以自动添加缺失的氢原子。
特点:
- 力场依赖:加氢基于所选力场(如ff19SB)的模板,氢原子类型与力场匹配。
- 简单直接:一条命令自动完成加氢、添加溶剂、离子等步骤。
- 默认质子化状态 :默认添加标准质子化状态(如Lys带正电、Glu带负电),但不自动优化His/Asn/Gln的取向。
典型命令(在tleap内):
source leaprc.protein.ff14SB # 加载力场
mol = loadpdb input.pdb # 加载PDB,自动加氢优点:
- 流程简单:适合结构规范、无需特殊处理的体系。
- 集成化:加氢与后续步骤(溶剂化、中和)一气呵成。
关键区别对比
| 方面 | pdb4amber + reduce | tleap 自动加氢 |
|---|---|---|
| 氢原子取向优化 | ✅ 通过Reduce算法优化(如His翻转、羟基取向) | ❌ 仅按模板添加,不优化 |
| 修复结构错误 | ✅ 自动修复残基/原子命名、缺失重原子 | ❌ 遇到错误可能直接报错 |
| 质子化状态控制 | ✅ 可通过pH值预测,并手动调整 | ⚠️ 仅默认状态,需手动修改残基名称(如HIE/HID) |
| 流程复杂度 | 多一步预处理,但更稳健 | 一步到位,但可能隐藏结构问题 |
| 适用场景 | 晶体结构分辨率低、有命名问题、需精确质子化状态 | 结构规范、无需特殊处理的体系 |
3. 推荐工作流程
结合两者优势的最佳实践:
-
初始检查:
pdb4amber -i 7SI1_prep.pdb -o 7SI1_clean_noH.pdb --nohyd # 检查问题但不加氢 -
修复并加氢(如需优化氢键):
pdb4amber -i 7SI1_clean_noH.pdb -o 7SI1_clean.pdb --reduce # 加氢并优化 -
手动调整(如有必要):
- 检查
clean_H.pdb中His、Asn、Gln的取向,用Chimera/VMD确认。 - 修改残基名称(如HIP/HID/HIE)以匹配质子化状态。
- 检查
-
进入tleap:
source leaprc.protein.ff19SB mol = loadpdb 7SI1_clean_H.pdb # 此时tleap不会重复加氢(因PDB中已含氢) solvateBox mol TIP3PBOX 10.0 addions mol Na+ 0 saveamberparm mol 7SI1_solv.prmtop 7SI1_solv.inpcrd
⚠️ 注意事项
- 氢原子重复问题 :若PDB已含氢,tleap默认不再添加,但会检查是否与力场匹配。
- His质子化 :晶体结构中的His通常为中性(HID/HIE),但需根据pH决定是否质子化(HIP)。pdb4amber的
--reduce可根据pH预测,而tleap需手动指定残基名。 - 氢原子命名:Amber力场对氢原子名称有严格规定(如HG1、HH22),pdb4amber可转换至正确命名。
小结建议
- 优先使用pdb4amber --reduce:尤其对晶体结构,可避免后续因氢原子问题导致的能量异常。
- 直接使用tleap:仅当PDB文件来源可靠(如经MD预处理)、无需优化氢键时。
- 始终可视化检查:无论用哪种方法,用VMD/Chimera检查加氢后的结构是否合理(特别是带电残基和氢键网络)。
✅ 六、验证与质检
-
使用parmed检查:parmed 7SI1_solv.prmtop << EOF
printDetails :HIE
EOF -
使用PyMol等查看溶液体系:
pymol 7SI1_solv.pdb
可以看到,溶液边界将蛋白部分完美包裹。需要确保蛋白不跨越周期性边界,通过solvateBox边界调节,一般10-12Å足够,太高会导致体系太大,运行效率降低。

✅ 七、体系准备与MD
后续工作按照蛋白溶液体系模拟即可。
可以参考系列博文:
大体流程如下分阶段准备体系:
-
仅约束溶质,优化溶剂(500-1000步)
-
全系统最小化(无约束)
-
逐步升温(NVT系综,100K → 300K)
-
密度平衡(NPT系综,调整盒子尺寸)
-
生产模拟(NPT或NVE)
本文介绍Amber用于分子动力学模拟输入文件的准备:蛋白结构准备,一般流程和关键注意事项。