**截止2025年12月,RCSB PDB数据库中记录了超过20万条通过X-ray获得的晶体结构,**最优分辨率为0.48 Å。
在分子动力学(MD)模拟中,X 射线晶体结构 是最常用的起始构象来源。然而,并非所有 PDB 中的 X-ray 结构都适合直接用于 MD------分辨率、晶体堆积效应、缺失区域、质子化状态等因素会显著影响模拟的可靠性。
以下是 专为 MD 模拟设计的 X-ray 结构选用与评估指南。
一、X-ray 结构用于 MD 的核心挑战
| 问题 | 对 MD 的影响 |
|---|---|
| 无氢原子 | 需正确添加 H,否则静电/氢键错误 |
| 晶体堆积伪影 | 表面 loop 可能被"压扁",不代表溶液构象 |
| 缺失残基/原子 | 主链断裂 → 力场崩溃 |
| 非生理 pH/突变体 | 质子化状态或关键残基异常 |
| 配体/辅因子建模错误 | 虚假相互作用 |
📌 关键认知 :
X-ray 结构是晶体中的"冻结"状态,不等于溶液中的动态构象。需谨慎评估其生物学相关性。
X-ray 结构解析相关参数,如何深刻的影响MD?总结如下:
| 特性 | 说明 | 对MD的影响 |
| 分辨率 | 通常1.0-3.0 Å | 原子位置精确,适合MD |
| 温度因子(B-factor) | 反映原子振动/无序 | 识别柔性/不确定区域 |
| 电子密度 | 实验观测基础 | 验证模型可靠性 |
|---|
二、X-ray 结构选用标准(按优先级排序)
晶体学统计指标记录的来源:
# PDB文件头关键字段解析
grep -E "REMARK 2|REMARK 3" 1abc.pdb
REMARK 2 RESOLUTION. 1.85 ANGSTROMS ← 分辨率
REMARK 3 R VALUE : 0.178 ← R-work
REMARK 3 FREE R VALUE: 0.215 ← R-free
REMARK 3 R FREE/R WORK RATIO: 1.208 ← 比值1. 分辨率(Resolution)
- ≤ 1.8 Å: 理想(侧链 rotamer、水分子可靠)
- 1.8--2.5 Å:良好(主链和大部分侧链可信)
- 2.5--3.0 Å:可用(需验证关键区域)
- > 3.0 Å: ❌ 避免用于全原子 MD(除非研究主链动力学)
💡 注意:高分辨率 ≠ 高质量模型!需结合 R-factors 和几何验证。
2. R-factors(数据拟合质量)
| 指标 | 好结构阈值 | 说明 |
|---|---|---|
| R-work | < 0.20 | 模型对衍射数据的拟合度 |
| R-free | < 0.25 | 防过拟合的关键指标(越接近 R-work 越好) |
📌 警惕:R-free > R-work + 0.05 → 可能过拟合。
3. 电子密度图质量
从PDB下载2Fo-Fc和Fo-Fc密度图(.ccp4或.mrc格式)或使用EDS(Electron Density Server)。
**2Fo--Fc map:**观测振幅(Fo)与计算振幅(Fc)的加权组合,显示主链和侧链是否被连续、合理的电子密度覆盖。
Fo--Fc map(差值图):Fo -- Fc 的残差密度,揭示模型中缺失/多余的部分(正密度 = 缺失原子;负密度 = 多余原子)。
| 项目 | 合格标准 |
|---|---|
| 主链 2Fo--Fc 覆盖率 | >95% 残基被 ≥1.0σ 密度包裹 |
| 最大 Fo--Fc 峰 | <3.0σ(活性位点除外) |
| Ramachandran 异常 | <1% 残基在 outlier 区 |
| 配体密度 | 清晰连续,无 >3σ 未解释密度 |
| 分辨率 | ≤2.5 Å(越低越好) |
密度图质量评估:
| 特征 | 含义 | 处理建议 |
|---|---|---|
| 主链密度断裂 | 无序或建模错误 | 检查B因子,考虑删除 |
| 侧链无密度 | 高柔性或多构象 | 保留最占据构象或删除 |
| 配体密度不完整 | 部分占据或错误建模 | 删除或重新拟合 |
| 水分子无球形密度 | 虚假水分子 | 删除 |
| 金属离子畸变密度 | 配位几何错误 | 修正配位或删除 |
4. 生物组装(Biological Assembly)
- 不要直接使用 Asymmetric Unit(AU)!
- 在 RCSB PDB 页面点击 "Download Files → Biological Assembly" 获取生理 oligomer。
- 原因:活性位点常位于亚基界面,AU 可能截断关键相互作用。
5. 配体与辅因子
- 优先选择 含天然配体/底物/抑制剂 的结构(PDB 中
HETATM); - 检查配体 B-factor:< 60 Ų 较可靠,> 80 Ų 可能位置不准;
- 避免使用 DMSO、PEG 等结晶试剂 作为功能配体。
6. 结构状态匹配研究目的
| 研究目的 | 结构选择要点 |
|---|---|
| apo蛋白动态 | 选择最高分辨率apo结构 |
| 配体结合机制 | 对比holo vs apo结构,选有配体的 |
| 酶催化机制 | 选择底物/过渡态类似物/产物复合物 |
| 别构调节 | 选择活性态 vs 非活性态构象 |
| 突变效应 | 优先选野生型,或相同突变体 |
| pH效应 | 注意结晶pH(常4.5-8.5),匹配生理条件 |
三、X-ray 结构质量评估(MD 前必做)
步骤 1:几何与立体化学验证(MolProbity)
使用 MolProbity 或 phenix.molprobity:
| 指标 | 优秀阈值(高分辨率结构) | 可接受阈值 |
|---|---|---|
| Ramachandran favored | > 98% | > 95% |
| Rotamer outliers | < 0.5% | < 1.0% |
| Clashscore | < 5 | < 10 |
⚠️ 特别关注:活性位点残基是否在 Ramachandran 允许区?
步骤 2:缺失区域处理
- 查看 PDB REMARK 465:列出缺失的残基;
- 短缺失(1--5 residues) :用 MODELLER 、Rosetta 或 PDBFixer 补全;
- 长缺失/柔性 loop:若不在功能区,可截断;否则需谨慎建模并验证。
- 不同来源PDB结构中残基缺失情况的处理总结:
| 方法 | 缺失特点 | 处理优势 | 处理策略 |
|---|---|---|---|
| X-ray | 柔性区无密度 | 高分辨区精确 | Modeller/ROSETTA填补,密度引导 |
| Cryo-EM | 局部分辨率差异 | 大分子完整 | 低分辨区删除或柔性拟合(MDFF) |
| NMR | 系综显示多态 | 动态信息 | 选取最完整模型,或删除无序区 |
| AlphaFold | 预测完整序列 | 无缺失 | 直接使用,或融合实验高置信区 |
| 整合结构生物学 | 多方法互补 | 交叉验证 | 使用SAS/EM/X-ray共同约束建模 |
💡 MD 提示:未补全的断裂主链会在能量最小化时崩溃。
步骤 3:质子化状态校正(pH 7.4)
X-ray 结构不含氢,且残基命名可能不反映真实质子化:
| 残基 | 默认状态(pH 7.4) | AMBER 命名 | 是否需手动干预? |
|---|---|---|---|
| Asp/Glu | 去质子化(--COO⁻) | ASP/GLU | ✅ 若在疏水口袋(如酶活性位点)→ 改为 ASH/GLH |
| His | 部分质子化 | HID(默认) | ✅ 必须根据氢键环境指定 HID/HIE/HIP |
| Cys | 质子化(--SH) | CYS | ✅ 若形成二硫键 → CYX;若配位金属 → 可能去质子化 |
| Lys/Arg | 质子化 | LYS/ARG | ❌ 通常无需改 |
🔧 工具推荐:
- H++ Server:预测 pKa 并输出质子化 PDB;
- propKa:命令行 pKa 预测;
- 手动重命名:将 HIS → HID/HIE,ASP → ASH 等。
步骤 4:水分子取舍
- 保留 :
- 活性位点水,尤其是结构水(介导配体-蛋白氢键);
- 金属配位水;
- B-factor < 40 Ų 且有氢键网络支持。
- 删除 :
- 表面水(B-factor > 60);
- 无明确功能的水(MD 会重新溶剂化)。
在蛋白质结构(尤其是来自 X 射线晶体学 或 冷冻电镜 的 PDB 文件)中,水分子通常以 HOH、WAT 或 H2O 残基形式存在。根据其物理化学角色和稳定性,可将这些水分以下几类:
| 类别 | 功能角色 | MD保留 | 识别特征 | 典型示例 |
|---|---|---|---|---|
| 结构水 (Structural) | 稳定二级/三级结构 | ✅ | B-factor<30, 3-4氢键, 同源保守 | 丝氨酸蛋白酶氧阴离子洞 |
| 配位水 (Coordinating) | 金属离子配位/催化 | ✅ | M-O距离2.0-2.5Å, 配位几何 | 锌指蛋白Zn²⁺-OH₂, 血红蛋白Fe²⁺-O₂ |
| 埋藏水 (Buried/Internal) | 填充疏水核心空腔 | ✅ | 完全包埋, 无溶剂接触, B-factor<25 | 蛋白内部空腔水, 促进折叠 |
| 通道水 (Channel/Pore) | 离子/分子传输链 | ✅ | 孔道连续性, 选择性滤器 | 水通道蛋白AQP, 钾离子通道 |
| 界面水 (Interface) | 介导蛋白-蛋白/DNA识别 | ⚠️ 视情况 | 界面凹槽, 部分保守 | 抗原-抗体界面, 转录因子-DNA |
| 变构水 (Allosteric) | 长程信号传递网络 | ✅ | 功能态差异, WaterMap高能量 | 血红蛋白T↔R态, GPCR激活 |
| 催化水 (Catalytic) | 直接参与化学反应 | ✅ | 亲核攻击, 质子穿梭 | 蛋白酶催化三联体, 碳酸酐酶 |
| 桥接水 (Bridging) | 连接蛋白-配体/辅因子 | ✅ | 双重氢键, 药物设计关键 | 激酶-抑制剂, 蛋白酶-底物 |
具有重要功能的水分子,如结构水、配位水、变构水和通道水等等,需要,首先按功能分类,结合 B-factor/密度/保守性判断,然后有依据地保留关键水。
| 方法 | 水分子可靠性 | 建议 |
|---|---|---|
| X-ray(≤1.8 Å) | 高(可分辨水) | 仔细筛选结构水 |
| X-ray(>2.5 Å) | 低(水位置不准) | 通常全部删除,仅靠 MD 重溶剂化 |
| Cryo-EM(≤2.5 Å) | 中(近年高分辨 Cryo-EM 可见水) | 仅保留密度连续、B-factor 低的水 |
| NMR | 部分可靠(通过 NOE 推断) | 通常不建模水,MD 中重新添加 |
💡 经验法则 :
分辨率 > 2.5 Å 时,不要信任任何水的位置!
四、常见陷阱与规避
| 陷阱 | 后果 | 规避方法 |
|---|---|---|
| 使用 AU 而非 biological assembly | 缺失关键界面 | 下载 biological assembly |
| 忽略活性位点 ASP 质子化 | 催化机制错误 | 用 H++/propKa 预测 |
| 保留高 B-factor 配体 | 虚假结合模式 | 检查配体 B-factor 和密度(如有 2Fo-Fc) |
| 不补全缺失 loop | MD 崩溃 | 用 PDBFixer/MODELLER 补全 |
| 直接用 tleap 加氢而不修正 HIS | 氢键网络错误 | 先重命名 HIS 为 HID/HIE |
五、X-ray 结构用于 MD 的 Checklist
实用评估清单
| 检查项 | 工具/方法 | 通过标准 |
|---|---|---|
| 分辨率 | PDB头 | ≤ 2.5 Å |
| R-free | PDB头 | < 0.26 |
| 密度图质量 | CCP4/PyMOL | 主链连续,侧链有密度 |
| Ramachandran | MolProbity | > 95% favored |
| 立体化学冲突 | MolProbity | Clashscore < 20 |
| B因子分布 | 自写脚本 | 无 >80 Ų异常峰 |
| 金属配位 | PyMOL测量 | 符合已知几何 |
| 配体合理性 | Ligand Expo | 键长/键角正常 |
| 晶格接触 | PISA | 活性位点无接触 |
| 水分子筛选 | B因子分析 | 保留B<30的结构水 |
必做项:
- 分辨率 ≤ 2.5 Å(功能研究)
- 使用 Biological Assembly
- MolProbity 报告良好(Ramachandran > 95%)
- 补全缺失残基
- 手动修正 HIS/ASP/GLU 质子化状态
- 保留关键水分子,删除表面水
- 用
pdb4amber清洗后交tleap处理
禁止项:
- 使用 R-free > 0.30 的结构;
- 不检查就运行含配体的 MD;
- 忽略晶体突变(如 Cys→Ser)对功能的影响。
六、MD模拟前的预处理流程
1. 标准预处理流程
下载PDB结构,使用pdb4amber初步处理,tleap构建体系。
# Step 1: 下载并基础清理
下载X-ray PDB结构去除杂原子(可选)
# Step 2: pdb4amber处理
pdb4amber -i PDBID.pdb -o PDBID_processed.pdb \
--reduce \ # 加氢
--dry \ # 去除水(可选,保留关键水需手动)
--most-populous \ # 选择最占据构象
--keep-heterogens # 保留配体/离子
# Step 3: tleap构建拓扑
cat > leap.in << EOF
source leaprc.protein.ff19SB
source leaprc.water.tip3p
mol = loadPDB PDBID_filtered.pdb
solvateBox mol TIP3PBOX 12.0
addIons mol Na+ Cl- 0.15
saveAmberParm mol PDBID.prmtop PDBID.inpcrd
quit
EOF
tleap -f leap.in2. 严格能量最小化
# 两阶段最小化应对晶体学应力
# step1_min.in: 约束重原子,优化氢
&cntrl
imin=1, maxcyc=5000, ncyc=2500,
ntb=1, cut=10.0,
ntr=1, restraint_wt=50.0, # 强约束
restraintmask='!@H=',
/
# step2_min.in: 全系统最小化
&cntrl
imin=1, maxcyc=10000, ncyc=5000,
ntb=1, cut=10.0,
ntr=0, # 无约束
/依次运行:
pmemd -O -i step1_min.in -o step1_min.out -p PDBID.prmtop -c PDBID.inpcrd -r step1_min.inpcrd -x step1_min.nc -ref PDBID.inpcrd
pmemd -O -i step2_min.in -o step2_min.out -p PDBID.prmtop -c step1_min.inpcrd -r step2_min.inpcrd -x step2_min.nc -ref step1_min.inpcrd获得经过能量最小化后的体系。可进一步通过体系加热(Heating)、密度平衡(Equilibration)之后,进行一定时长的正式MD,获得实验温度、密度和大气压下的模拟轨迹(traj.nc)。具体操作可参考博文:Amber分子动力学模拟2: 蛋白水溶液体系的模拟
七、与MD结果的一致性验证
1. 晶体学验证模拟
# 从MD轨迹计算模拟vs晶体结构的RMSD
cpptraj -p prmtop -y traj.nc << EOF
reference crystal.pdb
rms reference :1-300@CA out rmsd_to_crystal.dat
EOF
# 预期:
# 初始RMSD ~ 1.0-2.0 Å(晶体优化后)
# 平衡后RMSD ~ 1.5-3.0 Å(天然波动)
# 若RMSD > 4.0 Å:检查是否构象转变或结构问题2. B因子对比
# 计算MD的RMSF并与晶体B因子对比
cpptraj -p prmtop -y traj.nc << EOF
atomicfluct :1-300@CA out md_rmsf.dat byres
EOF
# 相关性分析(Python)
# 高相关性(R > 0.6)表明MD复现了晶体柔性模式
# 低相关性可能提示:
# - 力场问题
# - 晶体堆积效应
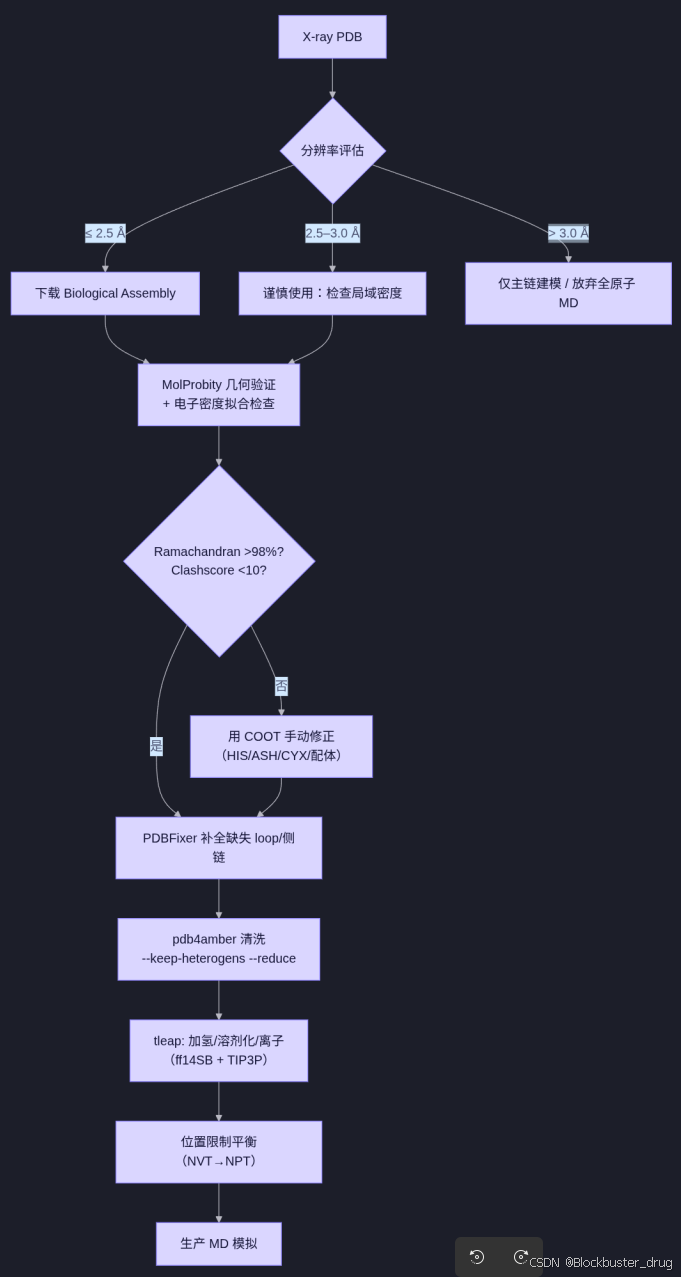
# - 时间尺度不足八、决策流程
九、常见问题处理
Q: 高分辨率但R-free异常高?
可能原因:
- 过度拟合(数据/参数比 < 2)
- 各向异性校正不当
- 双晶孪生未处理
建议:检查原始论文,谨慎使用Q: 活性位点有未解释密度?
可能为:
- 未建模的配体/离子
- 部分占据的底物
- 冷冻保护剂(甘油、PEG)
建议:查看Fo-Fc图,必要时删除可疑原子Q: 多个晶体学构象(altloc A/B)?
# 选择策略:
# 1. 选择占据率高的(>0.6)
# 2. 选择与研究目的更相关的(如开放/闭合态)
# 3. 分别模拟两个构象,对比结果
# pdb4amber自动选择最占据构象
# 或手动编辑PDB保留特定altloc
grep -v " B " structure.pdb > structure_altA.pdb # 删除B构象X-ray结构是MD模拟最常用、最可靠的起始点,但必须经过严格的质量评估,特别是电子密度验证 和B因子分析 。高分辨率(<2.0 Å)配合良好的R-free(<0.22)和清晰的密度图,是MD成功的最佳保障。高质量的起始结构是可靠 MD 模拟的基石。花 30 分钟评估,可避免数天的无效计算。