本来是想写GW计算流程,并且这部分其实在年前就已经跑过了,但是也仅限跑过,结果一直不尽人意,然后就是各种各样的事情耽误,总之决定先不搞GW了,毕竟这玩意计算成本摆在这,也不算是入门学习的东西。
那我们就先搞一遍电子结构分析和光学分析
我的知乎:
瞎某某Blinder![]() https://www.zhihu.com/people/jue-shi-tian-cai-xia-mou-mou
https://www.zhihu.com/people/jue-shi-tian-cai-xia-mou-mou
我的CSDN:
https://blog.csdn.net/Blindbaby123![]() https://blog.csdn.net/Blindbaby123
https://blog.csdn.net/Blindbaby123
我的B站https://space.bilibili.com/228111742![]() https://space.bilibili.com/228111742
https://space.bilibili.com/228111742
1.准备工作
我们在第2章的时候讨论过能带的计算,我们是先进行结构优化,然后计算SCF,然后计算能带
DFT学习记录2:VASP能带计算全流程![]() https://zhuanlan.zhihu.com/p/2003150720363435306
https://zhuanlan.zhihu.com/p/2003150720363435306
电子结构分析其实和这个差不多,只不过我们这次多加一个对态密度的讨论
光学分析则是将最后一步的能带计算修改为介电函数的计算,再通过介电函数推导吸收谱、折射率、反射率等光学性质
我们还是以mp-661AlN为例,但是我们这次省略掉结构优化这一步,我们关注比较重要的SCF之后的步骤首先是它的POSCAR
python
Al2 N2
1.0
3.1285879612 0.0000000000 0.0000000000
-1.5642940998 2.7094368588 0.0000000000
0.0000000000 0.0000000000 5.0169548988
Al N
2 2
Direct
0.666667000 0.333333000 0.499287000
0.333333000 0.666667000 0.999287000
0.666667000 0.333333000 0.880713000
0.333333000 0.666667000 0.380713000然后要生成KPOINTS和POTCAR,使用vaspkit,1,102,有以下几个选项,我们在之前没有解释清楚,我们这里补充一下
python
1) Monkhorst-Pack Scheme
2) Gamma Scheme
3) Irreducible K-Points with Gamma Scheme
##
1.首先 Monkhorst-Pack Scheme是最常用的,适用对称性较低的体系,基本可以进行所有体系的高精度计算
优点:普适性强,精度稳定;缺点:k 点数量相对较多,计算成本稍高
2.接下来Gamma Scheme适用于高对称性体系
优点:计算效率略高于 MP(对称性利用更充分),Gamma 点天然包含;缺点:低对称体系下采样均匀性不如 MP
3.最后Irreducible K-Points with Gamma Scheme(对称约化 + Gamma 中心)是先按 Gamma Scheme 生成网格,再利用晶体对称性约化掉等价的 k 点,只保留布里渊区的不可约 k 点,适用于高对称性体系与大规模计算
优点:k 点数量最少,计算速度最快,精度与完整 Gamma 网格一致;缺点:不适合对称性极低的体系(约化后可能丢失关键采样点
##当你不知道用什么的时候直接无脑1就行了,精度0.04就够,完成之后编写INCAR
python
# ========== 任务基本信息 ==========
SYSTEM = AlN-SCF # 计算任务命名,方便识别
ISTART = 0 # 0=从头开始计算(首次SCF必选);1=读取WAVECAR继续
ICHARG = 2 # 2=根据原子位置生成初始电荷密度(SCF初始态);11=非自洽
# ========== 精度与收敛 ==========
ENCUT = 500 # 平面波截断能(eV),Al/N的PAW赝势建议≥450,500兼顾精度/速度
ISMEAR = 0 # 0=Gaussian展宽(半导体/绝缘体常用);-5=四面体积分(非自洽用)
SIGMA = 0.05 # 展宽宽度(eV),半导体选0.05足够,越小越精准但耗时
EDIFF = 1E-8 # 电子步收敛判据(eV/atom),1e-8保证SCF高度收敛
EDIFFG = -1E-2 # 离子步收敛判据(仅NSW>0时生效),负号表示以力为判据(eV/Å)
NSW = 0 # 离子弛豫步数,0=不弛豫(SCF仅算电子)
IBRION = -1 # 离子弛豫算法,-1=不弛豫
# ========== 电子结构相关 ==========
ISPIN = 1 # 1=非自旋极化(AlN无磁性,无需自旋);2=自旋极化
LORBIT = 11 # 11=输出PROCAR(后续VASPKIT分析DOS/能带需要)
LREAL = .FALSE. # .FALSE.=全精度投影(高精度计算必选);.TRUE.=实空间近似(提速但降精度)
PREC = Accurate # 精度等级:Accurate(精准)> Normal(常规)> Low(快速)使用脚本启动vasp运算,运行之后拿到CHGCAR和WAVECAR即可
2.电子结构分析
我们新建一个文件夹,把POSCAR和POTCAR和上一步生成的CHGCAR与WAVECAR放进来,然后我们需要vaspkit帮我们生成一套新的KPOINTS用来进行计算电子结构,进入vaspkit使用3
python
301) 1D Structure
302) 2D Structure
303) Bulk Structure
304) K-Path for Wannier90 Code
305) K-Path for Phonopy Code
306) K-Path for CP2K Code
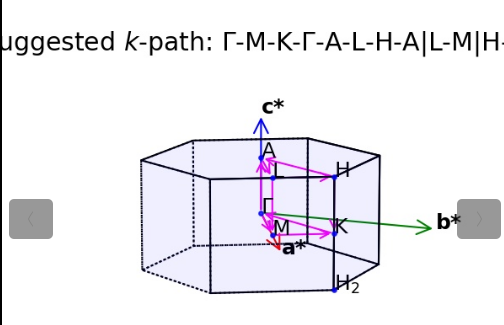
309) Visualize K-Path in First Brillouin Zone
##
常用的就是301 302 303,分别对应三个不同维度的材料,根据你的材料进行选择即可,我们的材料是三维,所以303
## 生成完成之后会给我们一张图和KPATH.in,把KPATH.in改成KPOINTS即可
最后写一下INCAR
python
# ========== 任务基本信息 ==========
SYSTEM = AlN-BAND-DOS # 任务命名:能带+态密度计算
ISTART = 1 # 1=读取上一步SCF的WAVECAR/CHGCAR继续
ICHARG = 11 # 11=固定电荷密度(非自洽计算,仅更新波函数)
# ========== 精度与收敛 ==========
ENCUT = 500 # 与SCF保持一致(截断能必须统一,否则电荷密度不匹配)
ISMEAR = 0 # Gaussian展宽(半导体/绝缘体通用,无k点限制)
SIGMA = 0.05 # 展宽宽度,0.05兼顾精度和速度(AlN建议0.02~0.05)
EDIFF = 1E-8 # 保持与SCF一致的高收敛精度
# ========== 电子结构相关 ==========
NSW = 0 # 不弛豫(非自洽仅算电子)
IBRION = -1 # 不弛豫
ISPIN = 1 # 非自旋极化(AlN无磁性)
LORBIT = 11 # 输出PROCAR,用于VASPKIT分析PDOS
NBANDS = 64 # 电子能带数,Al₂N₂共14个价电子,64足够覆盖导带(建议价带×3~5)
PREC = Accurate # 高精度保证能带/DOS准确性
# ========== DOS 专属参数 ==========
NEDOS = 2000 # DOS分点数,2000=分辨率高(曲线平滑)
EMIN = -20 # DOS能量范围下限(eV),覆盖Al/N的芯态+价态
EMAX = 20 # DOS能量范围上限(eV),覆盖导带然后运行脚本启动vasp运算,运算完成之后我们继续使用vaspkit,输入11进入态密度菜单,选择111,然后择 1) Energy in x-axis Versus TDOS in y-axis即可,生成TDOS.dat(总态密度核心数据文件),然后我们再次回到vaspkit,使用21,211,选择2 BAND + TDOS,他就会生成我们需要的图片和数据了,如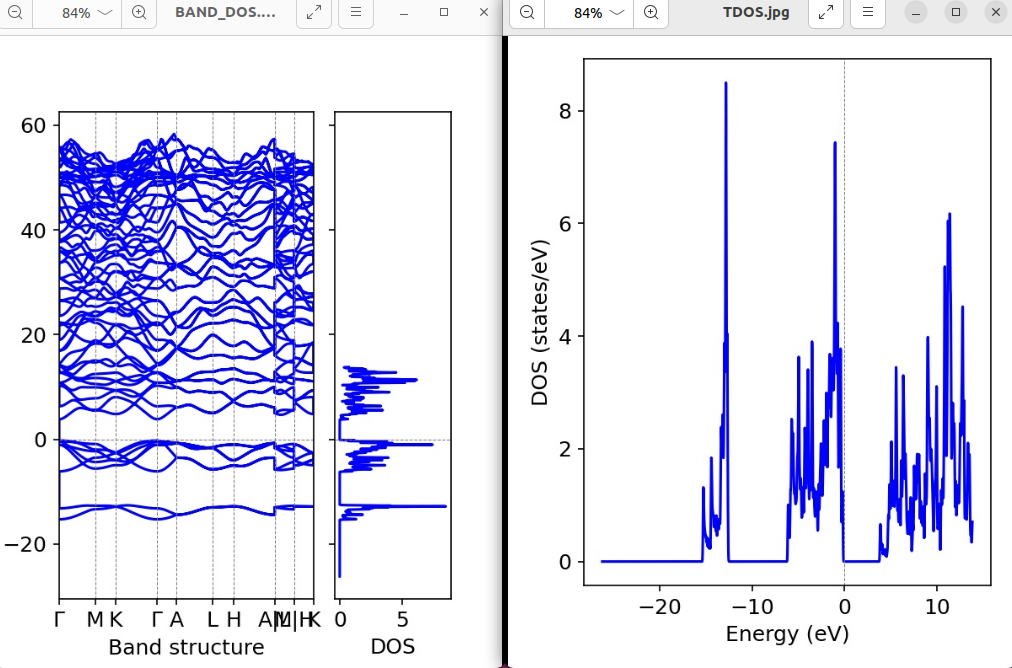
3.光学分析
光学分析反而简单一点,我们新建文件夹,把POSCAR POTCAR KPOINTS WAVECAR CHGCAR都放进来,然后写INCAR
python
# ========== 任务基本信息 ==========
SYSTEM = AlN-OPTIC # 任务命名:光学性质计算
ISTART = 1 # 读取SCF的CHGCAR/WAVECAR
ICHARG = 11 # 固定电荷密度(非自洽)
# ========== 精度与收敛 ==========
ENCUT = 500 # 与SCF一致
ISMEAR = 0 # Gaussian展宽(光学计算常用,平衡精度/速度)
SIGMA = 0.05 # 展宽宽度,0.05可抑制光学谱的噪声
EDIFF = 1E-8 # 高收敛精度保证介电函数准确
# ========== 电子结构相关 ==========
NSW = 0 # 不弛豫
IBRION = -1 # 不弛豫
ISPIN = 1 # 非自旋极化
PREC = Accurate # 光学性质对精度敏感,必须选Accurate
# ========== 光学性质专属参数 ==========
LOPTICS = .TRUE. # 开启光学性质计算(核心开关!)
LRPA = .TRUE. # .TRUE.=忽略电子-空穴相互作用(RPA近似,半导体光学计算常规设置)
NBANDS = 64 # 与能带计算一致,需覆盖足够多的导带(光学跃迁需要)
NEDOS = 2000 # 能量分点数,保证光学谱分辨率
EMIN = -20 # 能量范围下限
EMAX = 40 # 能量范围上限(光学跃迁需覆盖更高导带,比DOS多20eV)
# ========== 其他 ==========
# LPEAD = .TRUE. # 可选:包含电偶极矩的动量矩阵元(默认开启,无需修改)
# EPSILON = 1E-8 # 介电函数计算的收敛判据(默认足够)然后启动计算脚本运行vasp,等待计算完毕即可
进入vaspkit,输入71进入光学部分
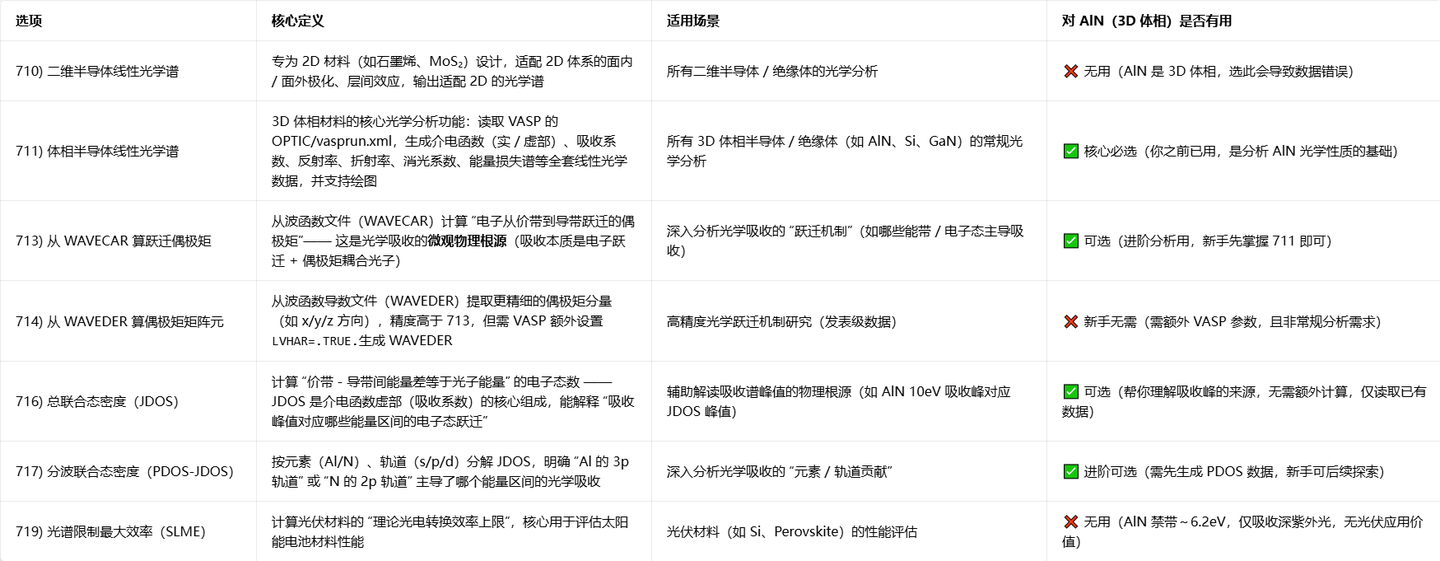
根据图示,我们选择711即可,随后为
python
Which Energy Unit do You Want to Adopt?
1) eV
2) nm
3) THz
4) cm^-1根据需求选择即可,我们选择1
最后我们就可以选择想要研究的内容了,分别为吸收系数,折射率,反射率,能量损失谱
python
Which optical quantity do you want to plot?
1) Absorption
2) Refractive
3) Reflectivity
4) Energy Loss Spectrum
5) Reflectivity 如果我们选择1,则会生成

我们就拿到我们需要的数据结果啦
最后,使用pymatgen貌似也可以进行光学分析与电子结构分析,但是目前这里学的还不够深,我们后面再专门开一期来学习pymatgen吧