论文总结
1、提出单分支与双分支图注意力网络架构,用于DTI影像数据的精神分裂症分类任务。其中,双分支结构分别提取各向异性分数与纤维数量两类脑网络特征,在全局池化层后通过拼接融合,再输入多层感知机进行分类,实现对白质结构信息的联合建模。
2、利用偏最小二乘回归,将双分支网络中FA分支的节点重要性权重与艾伦人脑图谱的基因表达数据进行关联分析,识别出与关键脑区显著相关的基因,并进一步开展功能富集分析,揭示精神分裂症相关的突触相关通路。
3、双分支GAT模型在1507名多中心样本上取得73.79%的分类准确率,优于单分支与其他对比方法。识别出的关键脑区(如扣带回、丘脑、额上回)及富集基因通路(如突触前后膜、轴突)与精神分裂症的已知神经生物学基础高度一致,为白质异常与遗传机制的桥接提供了新证据。
摘要
精神分裂症(SZ)是一种严重的精神疾病。本研究结合扩散张量成像(DTI)数据结合图神经网络,区分单纯的精神分裂症患者与正常对照组(NC),展示了整合分数各向异性和纤维数脑网络特征的图神经网络的优异性能,在区分单向异性患者与神经多样性方面达到了73.79%的准确率。超越单纯的区分, 我们的研究深入探讨了利用白质脑网络特征通过可解释的模型分析和基因表达分析识别SZ患者的优势。 这些分析揭示了脑成像标志物与遗传生物标志物之间的复杂关系,为SZ的神经病理基础提供了新见解。总之,我们的发现强调了将图神经网络应用于多模态DTI数据,通过神经影像和遗传特征的综合分析,增强SZ检测的潜力。
引言
精神分裂症(SZ)是一种严重的精神疾病,影响全球超过2000万人。患者终生受幻觉、妄想和认知缺陷等严重症状折磨,严重影响社交和职业功能1--4。2019年全球疾病负担研究(GBD 2019)的估计显示,SZ仍影响全球人口的约0.32%,过去十年患病率变化极小5。然而,存在显著的地区差异,尤其受医疗服务可及性、城市化以及社会和环境压力因素的差异影响6。最新研究强调了环境风险因素的重要性日益增加,包括城市生活、迁徙以及早期生活对逆境的暴露7。这些因素进一步复杂化了SZ的临床表现和管理,因此及时识别尤为关键。然而,症状表现上的巨大异质性使得仅凭临床评估诊断面临挑战。神经影像学和遗传学的进步越来越多地揭示了该疾病的根本神经生物学机制,使得更针对性的诊断和治疗方法得以实现。因此,利用客观的神经影像诊断辅助工具提升SZ检测的重视日益增加8, 9。在这方面,神经影像技术如MRI已成为阐明SZ复杂神经解剖异常的宝贵工具。此外,近年来,将机器学习应用于神经影像数据显示出有望揭示区分SZ患者与健康对照的分布式结构和功能模式10。某些研究聚焦于单一神经影像成像(如结构性磁共振成像)的机器学习算法11。例如,结构性磁共振成像的应用已实现了~80%的分类准确率12。相比之下,其他人则探索了静息状态功能性MRI在训练多变量分类器、评估功能连接性属性以及跨样本泛化方面的潜力13。值得注意的是,利用上颞叶皮层的功能连接特征,成功识别了SZ患者,准确率达78.6%14。尽管大多数研究聚焦于灰质和功能异常,但越来越多的证据表明白质异常在SZ病理中起着关键作用15。此外,最新证据强调白质完整性在SZ病理中的关键作用,这一点最好通过扩散张量成像(DTI)指标(如分数各向异性(FA)和纤维数(FN)来捕捉16。FA测量水的定向扩散程度,间接评估白质微观结构完整性。同时,FN量化了纤维束的数量,反映了大脑区域之间的连接强度。关注FA和FN特征的动机在于需要探索SZ中的白质完整性。它对SZ脑网络的潜在破坏提供了互补的视角。通过分析这两个特征,我们旨在更全面地理解SZ的神经病理。聚焦这些白质特征有助于更深入地研究SZ特有的脑网络紊乱。虽然有些数据包含了带有白质特征的多模态数据,但这些通常是辅助分类变量。很少有研究单独研究白质网络进行分类,通常使用小型单一地点、单模态样本,从而限制了跨多样人群的推广性17--20。例如,当仅应用DTI数据时,SZ的分类准确率从之前的75.05%下降到~70%和60%的两个数据集15。此外,处理大规模数据集的复杂性和多样性进一步加剧了这些研究方法的不足21, 22。深度学习中的图神经网络能够从多模态神经成像数据中捕捉高维关系,并表现出更优异的性能;他们有能力克服这些挑战23--25。虽然专为白质网络检测SZ而设计的深度学习技术较为稀少,但其效果常因不同地点收集的数据差异显著而受限,影响模型的泛化性和稳健性。近年来,转录组学和基因组学的重大进展为SZ的遗传基础提供了更深入的见解。大型基因组学研究,如精神病学基因组学联盟的,发现了众多与SZ相关的遗传位点,强调了该疾病的多基因特性26。转录组学研究进一步揭示了与神经传导和突触可塑性相关的脑区基因表达改变,这可能促成神经影像学研究中的结构变化27。通过将基因组和转录组数据与神经影像学整合,近期研究开始识别影响白质完整性的基因网络,从而更全面地了解SZ的潜在病理28。这些进展强调了将遗传数据与影像标记结合的重要性,以加深对SZ的理解并提升诊断精度。因此,本研究旨在利用图神经网络和扩散MRI开发一个可靠的深度学习框架,以揭示SZ相关的白质生物标志物。具体来说,我们使用了一个包含>1500名中国参与者的大型多站点数据集,这使我们能够推导出一个稳健且可推广的模型。此外,通过图神经网络分析和模型解释,我们努力识别相关的可靠结构变化对SZ。为此,我们引入了一种新型双分支图神经网络(GNN)模型,利用DTI数据,特别是FA和FN特征,以增强SZ的分类。该方法不仅整合了关键的白质完整性和连接性指标,还通过先进的协调技术有效减轻了多站点数据变异的影响。此外,通过整合遗传分析,我们旨在阐明影像标志物与遗传生物标志物之间复杂的关系。综合来看,希望这项工作能够促进临床辅助诊断的提升,并实现对该精神疾病的更准确检测。
材料和方法
整体流程图
该方法论如图1所示,包含三个主要组成部分:SZ分类、脑成像生物标志物的识别和遗传生物标志物分析。具体操作步骤如下:(1)SZ分类:利用DTI获得的脑网络结构数据,特别是FA和FN指标,构建个体层面的图结构表示。SZ通过应用单分支和双分支GNN架构进行分类。(2)脑成像生物标志物的识别:对于每个个体层面的图结构,计算节点权重值,随后确定平均权重。这一综合评估使得识别出与SZ显著相关的脑成像生物标志物。(3)遗传生物标志物分析:采用偏最小二乘(PLS)回归法阐明权重基质与基因表达基质之间的关系。通过这种分析方法,研究了与已识别脑成像生物标志物关联的遗传生物标志物。
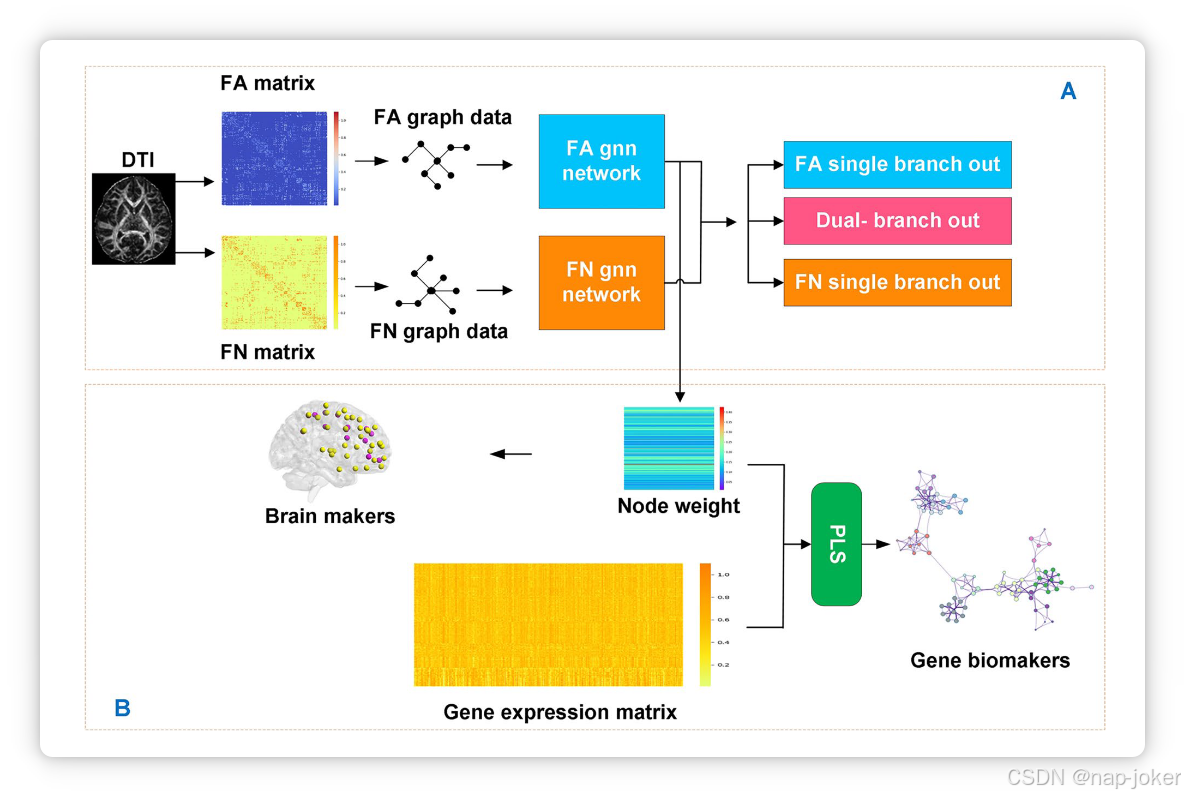
图1 整体流程图。SZ分类。脑网络结构数据来自扩散张量成像(DTI),并构建单个图结构数据。采用单分支和双分支GNN架构进行分类。B 脑成像生物标志物的鉴定,以及遗传生物标志物分析。基于双分支中FA分支对应平均脑面积得分,分析与SZ显著相关的脑影像生物标志物。PLS分析了重要权重矩阵与基因表达矩阵之间的关系,以及与已识别脑成像生物标志物相关的遗传生物标志物的探索。
参与者
该研究采用了来自中国各地的1507名参与者的多地点数据集。其中838人被诊断为SZ,669人为健康对照队列(见补充表S1)。SZ患者来自七家知名医院:北京大学第六医院、北京惠龙关医院、安徽医科大学、新乡医学院第二附属医院、广州精神病院、武汉大学人民医院和祖马丹精神病院。症状严重程度使用阳性和阴性综合征量表(见补充表S2)评估,该量表在MRI扫描后一周内对患者进行。
影像采集
数据集1由北京大学第六医院使用西门子TrioTim扫描仪获得。数据集2由北京惠龙关医院使用西门子TrioTim扫描仪获取。数据集3来源于安徽医科大学,使用GE医疗系统Signa HDxt扫描仪。数据集4由新乡医科大学第二附属医院通过两台设备获取。部分扩散加权成像(DWI)数据由西门子Verio扫描仪采集,部分DWI数据由GE医疗系统Signa HDxt扫描仪采集。数据集5在广州精神病院使用飞利浦扫描仪采集。数据集6在武汉大学人民医院使用GE医疗系统Signa HDxt扫描仪获取。数据集6在武汉大学人民医院使用GE医疗系统Signa HDxt扫描仪获取。数据集7在Zhumadian精神病院使用GE Medical Systems Signa HDxt扫描仪获得。所有DWI数据均采用单次自旋回声平面成像序列,参数见补充表S1
数据预处理
DTI数据集的预处理采用了Pipeline for Analyzing Brain DTI(PANDA)29,该软件专门用于扩散MRI分析。它包含了必要的处理步骤,包括头骨剥离、运动修正、涡流伪影校正以及扩散张量参数的计算。与先前建议一致30,本研究中每位受试者的DTI图像处理采用默认参数。预处理流程包括以下顺序步骤:(1)将图像从DICOM转换为NIFTI格式。(2)基于由T1加权图像生成的全脑遮罩,去除非脑组织。 (3)减轻头部运动并修正涡流伪影,以减少其对影像变形的影响。(4)利用线性最小二乘拟合方法计算并拟合每个体素的扩散张量度量。(5) 在原生扩散空间中,将单个T1加权结构图像与相应的非扩散加权(b = 0)图像共配。(6) 利用脑软件库的功能性磁共振成像,将转换后的结构图像与蒙特利尔神经研究所空间的标准模板对齐。在这些步骤中,涡流被认为是导致像形变的重要因素。应用仿射变换将DTI图像与T1加权图像对齐,有效减轻了头部运动和涡流的不良影响。排除未达到质量标准的数据后,保留了1507个可行样本。为促进脑区定位和分析,我们采用了脑网图谱31,该图谱包含246个子区域,包括210个皮层亚区和36个皮层下亚区,提供了全面的脑图谱框架。遵循先前研究建议32,集成于扩散工具包中的连续追踪纤维分配算法被应用于纤维牵引重建全脑连接组33。该算法涉及基于种子的双向跟踪,沿扩散张量的主特征向量进行跟踪,当传播线的挠度阈值超过45°时,纤维传播停止34。系统地识别每对脑网图谱区域间的白质连接,使得对应这些连接的纤维得以提取,建立了两个脑网络连接矩阵:平均FA矩阵和FN矩阵,这两个矩阵的维度为246×246。
种群分层与场地效应调整
鉴于本研究所用数据集包含来自七个不同地点的1507名参与者,存在因人口统计和扫描器相关差异而存在显著的人群分层风险。为减轻这些影响,我们采用了ComBat协调技术,该技术广泛应用于多位点神经影像数据,以减少位点特异性变异性,同时保持生物差异。ComBat方法在调整年龄和性别等协变量后应用,从而减少不同扫描仪或采集协议引入的非生物变异性影响。这一调整确保后续分析聚焦于SZ固有的神经生物学特征,而非由遗址差异引入的伪影。
在我们的研究中,ComBat协调技术应用于重建的FA和FN数据,而非原始数据,以减轻场地特异性变异性,同时保持生物变异性。我们使用场点变量作为批次标识符,并调整了年龄和性别等协变量,以更准确地消除批次效应,同时保留有意义的神经生物学差异。协调过程是在DTI数据预处理后进行的,预处理包括对涡流和运动伪影的修正,确保数据在应用ComBat调整前的一致性。
图数据的构建
图像预处理阶段完成后,为每位参与者生成两个大脑连接网络,每个网络大小为246×246。这些网络旨在表示Brainnetome图谱中划分的不同区域之间的互联。具体来说,网络由加权值组成,表示连接节点之间的平均FA或FN。由于FA和FN网络都类似于图结构化数据,我们分别独立构建了图数据。这些图数据集随后被用作模型的输入。在我们的模型中,每个样本对应两组图结构化数据,即FA图结构化数据和FN图结构化数据。在这些数据结构中,节点对应不同的大脑区域,总计246个节点。在构建节点特征时,我们使用FA网络的每一行作为对应节点的节点特征。这种方法有效地封装了特定节点与其余246个节点之间的连接性。同时,我们将网络视为邻接矩阵,其值用于确定对应节点之间的边连接。我们把阈值设为0。大于0的视为连通,小于或等于0的视为不连通。该方法论最终形成了相应的图结构化数据,如方程所示:其中G代表图结构化数据,V代表节点集合,E代表边集合,X代表节点特征集合。在构建FA网络的同时,也采用类似的程序构建FN网络的图数据,便于将其纳入图神经网络以进行分类。FN图结构化数据的表示方式如下:


基因数据和预处理
本研究采用了艾伦人脑图谱(AHBA)数据集,该数据集以其对整个人脑的全面高分辨率覆盖而闻名,作为基础资源。它包含了从六名成年脑供体提取的3702个不同空间样本的微阵列和RNA测序数据35。为提高全基因组与大脑成像数据的相关性,仅使用RNA测序数据。基因经过细致过滤和重新标记,利用Abagen(https://github.com/rmark ello/abagen)提供的工具。此外,AHBA供体的MRI图像被映射到脑网图谱中246个已划定的脑区。因此,成功推导出了维度为246×13561、值范围为0到1的基因表达矩阵,代表这13561个基因在246个脑区的表达水平。
图神经网络与分析
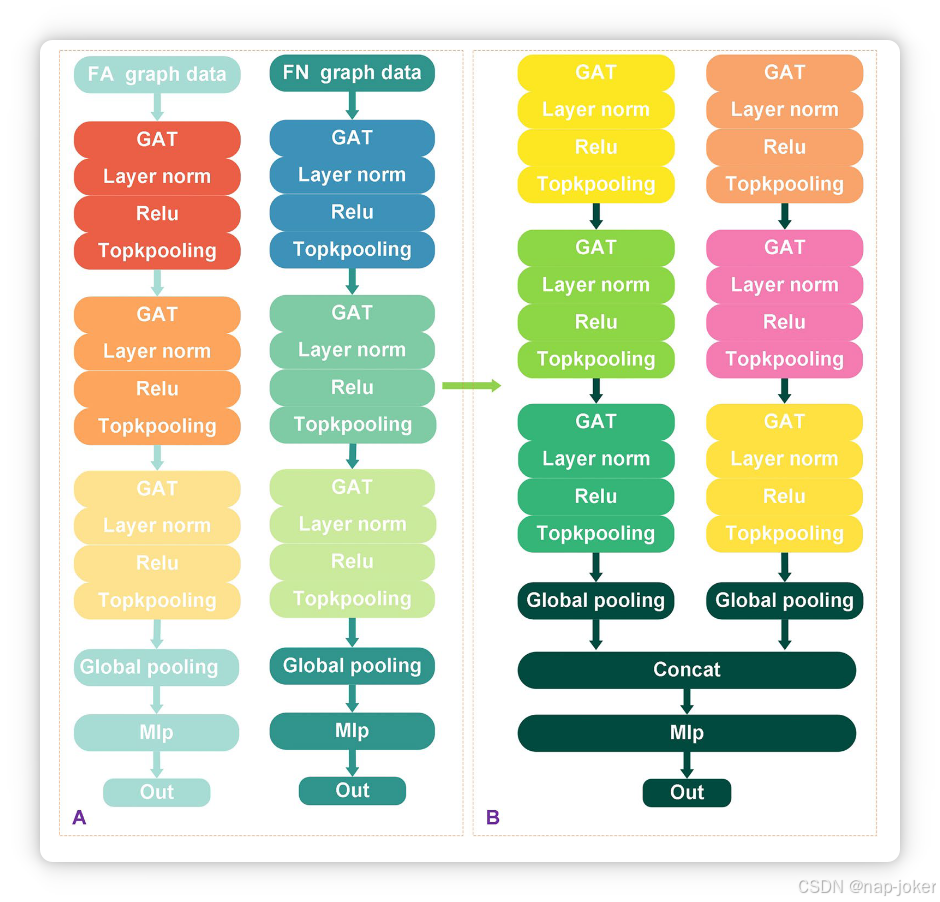
图2 图神经网络。A:单分支图神经网络。为FA和FN数据构建相同的网络结构:输入(GAT+层规范+Relu+Topkpooling)×3、全局池化、MLP和输出。B 双分支图神经网络。采用与FA和FN数据单分支相同的网络结构,并在全局池化后通过concat融合功能。
图注意力网络(GAT)是一种具有注意力机制的著名图神经网络架构,因其在多个领域的卓越表现而备受关注36--38。值得注意的是,其在MRI数据分类任务中的有效性已通过以往研究验证39。鉴于FA和FN数据的相关性,它们有效捕捉大脑的连接模式,我们选择从这些模态构建图结构化表示,作为对GAT的输入,用于我们的SZ分类任务。在此基础上,我们引入了一个双分支GAT网络,精心设计用于整合FA和FN数据以完成分类任务。同时,各分支仍保留使用相应FA或FN数据独立进行分类的能力,如图2所示。这种双分支架构允许利用两种模态的互补信息,同时评估它们各自的判别能力。GAT架构在捕捉不同脑区之间复杂非线性关系方面发挥着关键作用,得益于其多层设计,能够基于节点重要性动态学习特征。具体来说,GNN中的每一层都负责处理连通性数据,并通过迭代聚合和转换邻域信息来学习唯一的节点表示。第一层GNN捕捉了节点间最局部的连接,重点关注单个脑区内的直接关系。后续层次则建立在这些关系之上,这使得能够提取代表整个网络中越来越全局和抽象交互的高阶特征。GAT的多层结构帮助模型通过根据节点特征的连通性不同加权来学习非线性交互,增强捕捉兴趣区域(ROI)之间复杂功能关系的能力。在我们提出的网络架构中,我们对FA和FN图结构数据实现了单分支分类方法,采用相同的网络结构。整体网络设计包含三层GAT,每一层都能从图结构化数据中巧妙提取特征。值得注意的是,GAT 区别于传统的图卷积网络(GCN),不仅考虑邻居节点的信息,还考虑节点特征更新过程中节点间的权重,从而巧妙捕捉重要的节点间关系40。以下公式可用于表示具体的程序。GAT 的输入是一组节点特征:
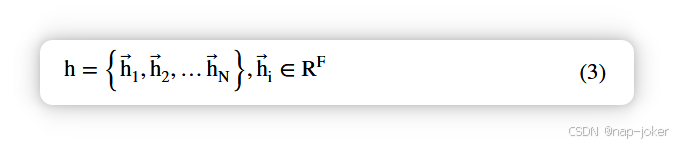
利用可学习权重矩阵W ∈ RF ×F,该矩阵应用到每个节点。共享注意力机制:一个 × RF → R,对节点进行自关注。可得节点j对节点i的特性重要性:


在每个图卷积层之后,我们应用了ReLU激活函数和层归一化技术。这有效解决了梯度爆炸或消失相关的问题,显著缩短了前馈神经网络的训练时间,并为分类过程提供了进一步的稳定性41。最后,应用多层感知器(MLP)对全局合并的特征进行分类。通过采用双分支网络,FA和FN数据被视为两个相关的子任务,并在并行分支中独立处理。对于每个分支,我们采用相同的图神经网络结构和数据输入,作为一个分支。具体来说,FA和FN分别使用三层GAT编码,使模型能够学习每种模态独有的特征。特征提取后,从每个分支学到的特征会经过全局池化层处理,该层降低了数据的维度,同时保留了每种模态中最具信息量的特征。然后通过串接方式融合这些特征,整合FA和FN提供的互补信息。这种串联表示捕捉了两种模态的独特性和共享性,增强了网络建模复杂关系的能力。融合后的特征随后会传递给MLP进行最终分类,为整合和利用每种模态的独特特性提供了稳健的机制。这一战略设计最大限度地利用了两个数据集的固有信息,结合FA的微观结构洞见与FN的连通性测量,有望提升分类准确性。此外,我们的网络架构具有可扩展性,熟练地使用相同的图神经网络结构,分别从FA和FN图结构化数据中提取特征。从每个分支获得的全局合并特征通过一个连接层有效地合并。最后,一个两层全联网网络基于合并特征进行分类,精确区分具有SZ和NC的个体。
可解释模型
鉴于GAT在分析复杂神经连接方面的前景,探索如何将白质的特定特征如FA和FN整合进该框架,以加深我们对SZ的理解变得至关重要。研究 影响的动机在SZ背景下,FA和FN在DTI(白质异常)中的重要特征,在于理解白质异常如何促成该疾病的病理。鉴于SZ表现为大脑多个区域广泛的结构和功能障碍,研究FA和FN的作用有助于了解与这些变化相关的具体连接模式。异常的FA值可能表明白质完整性受损,而FN特征则能揭示复杂的网络相互作用,这对理解SZ的神经生物学基础至关重要。关注这些特征可能有助于加深对白质连接性与SZ关系的理解,从而促进诊断和治疗方法的改进。在我们的研究中,通过实施GAT在SZ分类方面取得了有利结果。然而,GAT模型固有的非线性性使其内部动态和决策过程难以理解,因此模型成为难以解释和理解的"黑箱"42。为解决这一问题并降低复杂性,采用TopKpooling方法,通过根据学习到的注意力评分选择最重要的节点来提升可解释性。具体过程包括几个步骤:首先,我们创建一个246 * 1的可训练参数,并将节点特征246 * 246与可训练参数246 * 1相乘,得到更易管理的形式246 * 1。接下来,通过应用激活函数,我们为每个节点推导权重系数,从而形成大小为246 x 1的权重系数向量43, 44。随后,我们保留得分最高的前50%淋巴结,这使我们能够专注于对分类结果有显著贡献的脑区。通过检查这些选定的淋巴结(脑区),我们可以推断这些区域的连接性和完整性对于区分SZ患者与健康对照组尤为重要。这种选择性剪枝不仅降低了模型复杂度,还突出了作为潜在生物标志物的特定脑区,从而增强了我们发现的临床可解释性。具体程序可用以下公式表示:
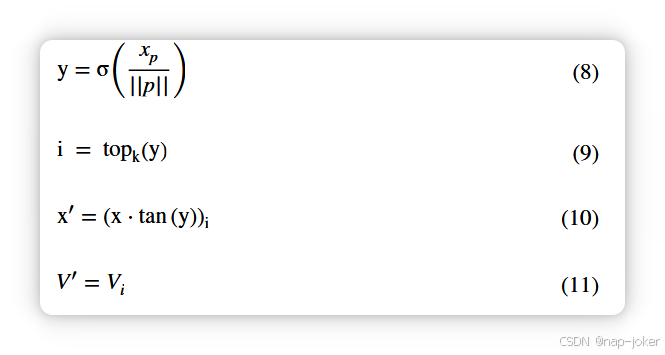
其中 x 表示节点特征 246 * 246,p 表示大小为 246 * 1 的可训练向量,x 乘以 p 得到 XP 为 246 * 1,Y 表示分数每个节点的K代表保留节点的数量,x'代表更新的特征,V'代表更新后的对应节点集合。通过采用这一方法,我们努力提升模型结果的透明度和可解释性,同时促进识别与SZ分类相关的关键脑区。
基因表达分析
本研究采用偏最小二乘法(PLS)研究遗传生物标志物与SZ脑图像权重之间的关系。其总体目标是发现基因表达矩阵与解释权重之间的回归关系。PLS回归是一种多变量统计方法,提供了一种有价值的方法来同时分析多个因变量。它在处理有限样本量和解决自变量多重共线性问题方面尤为有利。因此,PLS回归在该领域被广泛应用45--47。在我们的PLS分析中,基因表达数据作为自变量(预测变量),而从神经网络图中得出的脑区权重被视为因变量(反应变量)。该方法被用于分析基因表达与大脑投资回报率(ROI)之间的潜在关系,从而识别出与参与SZ的特定脑区密切相关的遗传标记。我们的分析显示,PLS1与预测变量的关联最强,节点权重累计解释方差为83.385%。为评估该关联是否超出偶然预期,我们进行了1000次自助重抽样迭代(P <0.05)48,并对每次重抽样重新计算PLS模型。自助分析显示,<1%的重抽样模型显示出由PLS1解释的方差,等于或大于原始模型(P <0.01),表明该关联具有统计学意义,不太可能是偶然发生。第一个潜在变量PLS1解释了节点权重方差的83.385%,这远高于偶然预期,从自助重抽样的P值<0.01中可见一斑。因此,PLS1上占较大权重的基因有可能与SZ脑成像标志物相关。虽然由于计算能力和数据可用性限制,我们未进行空间置换测试,但我们采用了1000次迭代的引导重采样作为替代方案。该方法提供了方差显著性的稳健估计。然而,未来的研究应考虑纳入空间置换测试,以更全面地直接处理空间自相关效应。
结果
分类表现能力
本研究的主要目标是评估利用DTI数据结合深度学习方法,对被诊断为SZ个体的大脑结构性改变进行分类和研究的潜力。我们的实验工作重点是评估FA和FN数据在单分支和双分支网络分类中的有效性。为了提升模型的泛化性,我们合并了7个不同地点的数据。随后,我们将合并数据划分为10个不重叠的部分,采用10折交叉验证方法进行实验。每个实验中,使用9个分区训练模型,1个分区留待验证。模型训练采用Adam优化算法,学习率为0.001,批处理规模为16。交叉熵损失函数成为分类过程的基础。此外,为了获得可靠的实验结果,我们应用了多种不同的随机种子,完全洗牌了训练集和测试集的数据分布,并重复了多次10折交叉验证实验。多次十折交叉验证的平均准确率误差在0.01以内,表明数据分布对本实验没有影响。多次10折交叉验证的准确性差异存在细微差异,通常保持在合理范围内。这表明数据分布对实验结果稳定性的影响有限。在评估分类模型时,我们应用了多种指标,包括准确率(ACC)、特异性(SPE)、敏感度(SEN)和F1分数。最终,我们的发现涵盖了单分支网络分类和双分支网络分类背景下,FA和FN数据的结果。详细数据(见补充表S3、S4和S5)已纳入,作为全面参考。
单一分支的分类结果
根据补充表S3的结果,我们提出的单分支GNN在FA数据集上的分类表现更优。学校平均ACC为72.59%,SEN为76.40%,SPE为67.83%,F1成绩为75.50%。这些结果突出单分支GNN在准确识别FA数据集中大多数样本并有效避免误分类方面的熟练能力。相比之下,补充表S4的结果显示,我们的单分支GNN在FN数据集上的分类表现略逊于FA数据集。其平均ACC为70.86%,SEN为73.19%,SPE为67.65%,F1得分为73.73%。这表明我们的单分支GNN在处理FN数据集样本时可能存在限制,表明需要进一步优化和改进。
双分支的分类结果
如补充表S5所述,我们提出的双分支GNN表现显著,平均ACC为73.79%,SEN为77.59%,SPE为70.11%,F1得分为75.96%。这些结果凸显了使用双分支结构相比单分支GNN在分类性能上的显著提升,表明该方法的有效性。高SEN意味着该模型在识别真正阳性SZ病例方面尤为有效,这对于确保个体获得及时且适当的治疗至关重要。与此同时,SPE数值表明模型有效减少了假阳性率,从而降低了健康个体误诊的可能性。这些指标共同标志着基于影像的SZ诊断可靠性取得的积极进展,暗示该方法具有潜在的临床价值。分类性能的提升强调了将白质脑网络特征(即FA和FN)与基于GNN的分析整合的重要性。这种整合具有显著的临床应用潜力,为临床医生提供一种非侵入性且客观的工具,帮助他们更准确地诊断SZ。通过结合这些互补特征,我们的双分支GNN不仅提升了分类的整体准确性,还通过强调与SZ病理相关的重要特征,提升了可解释性。基于这些发现,我们的双分支GNN在整合FA和FN的数据集上展现了最佳分类性能,这清楚地表明双分支网络有效利用了这两个数据集中的唯一特征。此外,在确立了双分支GNN模型在SZ分类中的稳健性后,我们接下来着手探索对分类结果有贡献的特定脑成像标志。通过识别和分析这些标志,我们试图从生物学角度解释SZ患者的结构变化,从而加深对该疾病背后神经生物学机制的理解。鉴于FA和FN数据集整合的优异表现,以及FA作为DTI图像中对应脑区指示的重要指标,我们也计划在未来研究中基于FA网络探索潜在的脑成像标志物和遗传生物标志物。最终,本研究旨在深化我们对SZ背后神经生物学机制的理解,重点是识别新的生物标志物,进一步提升诊断过程。
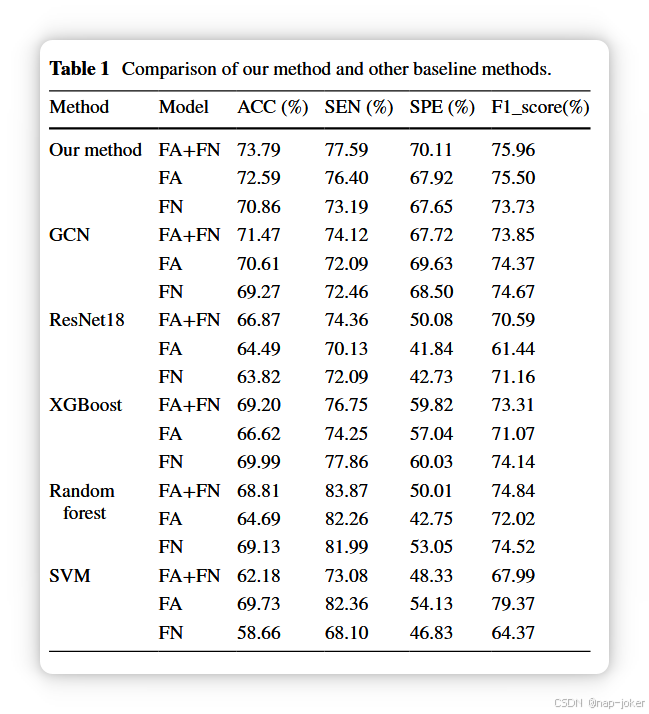
分类方法和其他方法比较
我们将模型与其他五种竞争方法进行比较:随机森林 49、支持向量机(SVM)50、XGBoost 51、ResNet 52和GCN 53。GCN采用与该实验方法相同的网络结构,SVM的惩罚参数为1.0,随机森林中有100个决策树,XGBoost中有100个加成回合,最大深度为6棵树。ResNet使用经典的ResNet18,但改变了输入大小并采用二进制分类。上述方法中的所有其他参数均为默认。我们评估了他们在多样化模态数据中的表现。我们使用F分数算法进行FA和FN矩阵的特征选择,特别选择前1000个特征作为机器学习方法的输入。在ResNet中,我们将矩阵视为二维图像,并作为网络输入。尽管FA和FN矩阵的表示方式不同(如特征向量、二维图像、图结构),但数据中的核心信息在所有模型中保持一致。我们对比较方法采用了10折交叉验证法,对于深度学习方法,我们采用了与本实验相同的训练策略。如表1和图3所示,我们的模型在ACC指标方面表现优于其他分类方法。此外,关于ACC、SPE、SEN、F1_score及曲线下面积,我们的模型在应用于FA + FN数据集时表现更优,超越了其他分类方法。这些结果展示了我们模型卓越的分类能力,有效区分异常样本并最大限度地降低假阳性率。
消除年龄、性别和地点差异的影响
为了确保我们发现的完整性,我们对样本进行了严格筛选,以应对年龄和性别差异等因素的潜在影响。最初,我们评估了两人的平均年龄差异NC组和SZ组,为减少年龄相关的混杂效应,我们从NC组中最年轻的31名参与者和SZ组中最年长的30名参与者中提取了样本。排除61个样本确保我们的分析聚焦于与SZ相关的脑结构变化最为明显且可比的核心年龄组。将参与者纳入极端年龄,可能会引入与正常发育或退行性变化相关的变异性,这些变化并非特有性特异性,可能会干扰我们的分析。通过排除这些异常值,我们旨在更好地隔离SZ特异性的大脑改变。随后,我们进行了统计检验,以确保NC组和SZ组人口统计变量的可比性。我们的分析,包括年龄的t检验和性别的χ2检验,显示两组之间无显著差异(P >0.05),再次确认了筛查后这些组的可比性。随后,我们使用精炼样本进行了10次交叉验证,确保实验设置的一致性。总结结果见表2。为了进一步解决不同采集点差异可能产生的偏差,我们对筛选样本应用了ComBat协调方法,详见54。该方法通过识别批次效应,通过站点变量标准化数据,同时调整年龄和性别等协变量,更准确地消除这些效应,同时保留有意义的生物学差异。该归一化过程对实验结果的影响详见表3。此外,对于我们的基线比较方法,我们应用了上述相同的预处理步骤;处理后的结果见补充表S6。经过仔细审查筛选数据,我们未发现年龄和性别间明显差异,进一步确认了我们筛查过程的稳健性。值得注意的是,实验结果与数据筛查前结果一致,FA、FN和FA+FN双分支变化较小。此外,ComBat应用于处理位点差异的结果也类似,强调了该方法在维护实验完整性方面的有效性。尽管FA、FN和双分支模型的性能指标存在细微差异,但这些变化均在可接受范围内,显示出我们的研究结果具有可靠性。基于我们表现最佳的融合双分支模型,我们得出结论,年龄和性别等人口统计变量以及场地变异的影响已被有效缓解。此外,采用10折交叉验证方法,合并7个站点样本,增强了模型的普适性和稳健性。
潜在的脑影像标记物
在我们的模型中,我们实现了TopKpooling方法,用于计算单个脑节点的评分,旨在提升分类性能并识别潜在的脑成像标志。通过评估对偶分支中的FA分支GNN,我们按降序组织了每个节点10折交叉验证的平均得分。值得注意的是,脑部前50个区域形成了明显的聚集,显著包括上额回、扣带回、中额回、丘脑、眼窝回和前楔形叶。图4所示的这些区域是SZ研究的关键关注领域,因为它们参与认知和情绪处理。例如,上额回与SZ患者工作记忆和执行功能缺陷相关,这与Ehrlich等(2012)和Mubarik等(2016)55, 56的发现一致。扣带回,尤其是其前部,被认为与情绪调节和社会认知有关,这与Thielen等人(2022)强调其在SZ相关认知障碍中的作用57相符。此外,丘脑在感觉整合和认知处理中起着核心作用,丘脑连接障碍是SZ病理的显著特征,Wang等人58报道了这一点。当前的研究结果强调了已识别的影像标志与成熟的SZ神经病理学之间的趋同,为这些区域的相关性提供了有力证据作为SZ的潜在生物标志物。上额回和扣带回等区域的双侧分布强调了它们在理解SZ症状背后神经机制中的重要性。
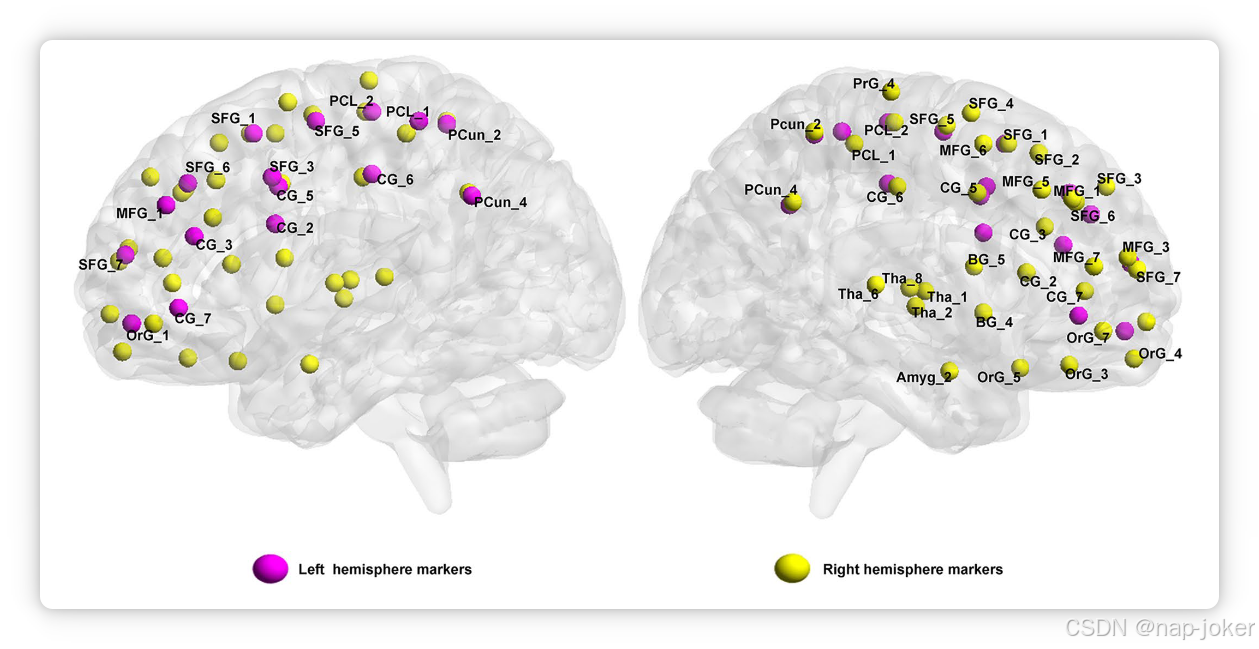
图4 与FA相关的潜在影像标志的可视化。对可能作为生物标志物的左右脑区分别标记。
潜在的基因生物标志物与富集途径
图5中,A部分展示了与PLS1基因相关的本体项的可视化(含pFDR <0.05)。可视化中每个圆的大小代表相关术语的普及率。B部分展示了富集网络,揭示了富集项群内及簇之间的相似性。圆形节点表示富集项,节点大小表示被包含基因的数量。此外,颜色编码方案突出群集身份,即同色节点属于同一群集59。在我们的遗传分析中,我们按降序排序了PLS1权重,识别出526个排名最高的和526个排名较低的基因(前10和后10个基因见补充表S7)。我们利用Metascape60对这些基因进行了全面分析,重点关注基因本体(GO)类别,如生物过程、细胞组分(CC)和分子功能(MF)。该分析使我们能够探索这些 GO 类别中丰富的术语,从而深入了解已识别基因的生物学作用。例如,CC中识别的GO术语包括突触膜和轴突成分等结构,而MF中则涵盖神经递质结合和离子通道活性。通过对这些丰富术语进行分类,我们详细介绍了其生物功能,突出它们对SZ病理学的潜在贡献。特别是在富集分析和富集条件调整(pFDR <0.05)并排除孤立簇后,我们发现与FA相关的前11大基因本体。著名术语包括"突触后"、"轴突"和"突触前"。这些发现为这些基因本体在SZ发病机制中的潜在作用提供了宝贵见解。
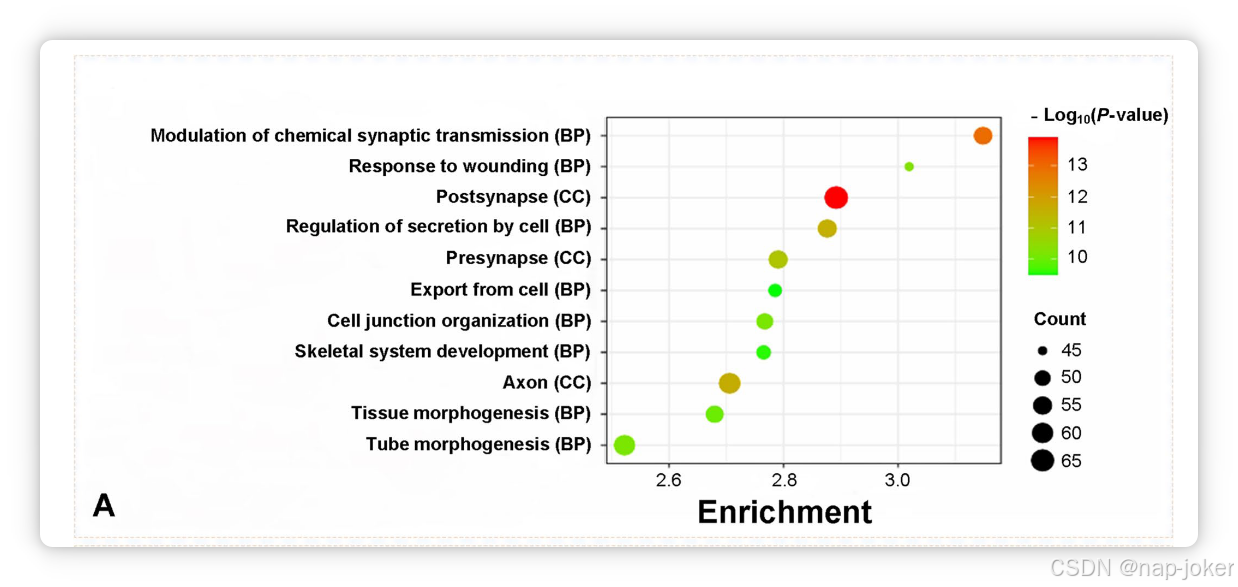
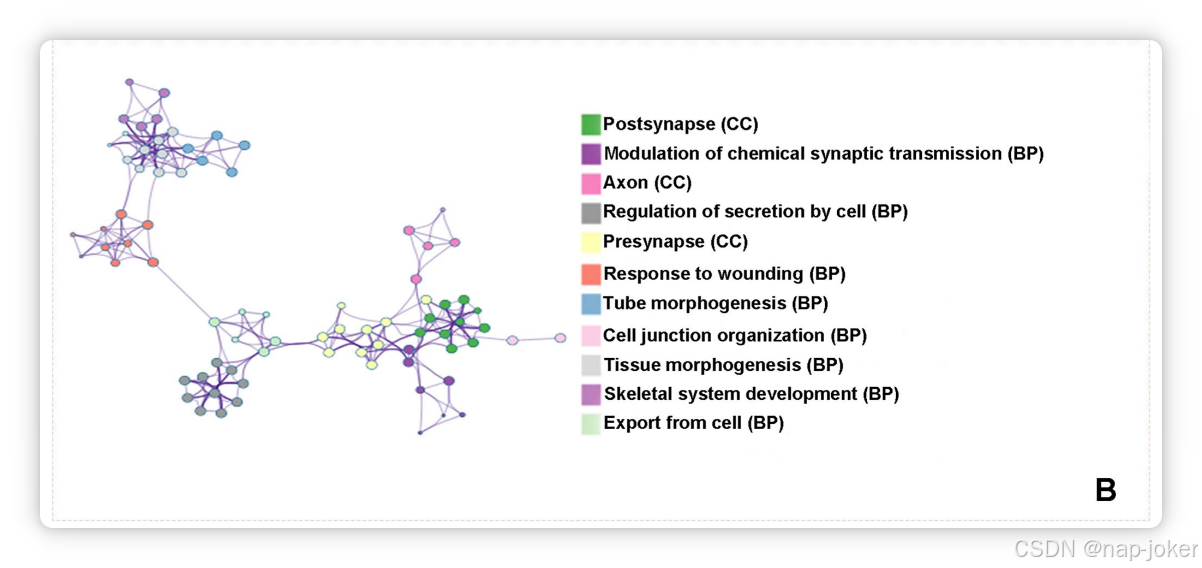
图5 基因转录本的功能富集。一个充实点泡泡。气泡颜色,即转换后的p值;气泡大小,即基因数量;x轴,浓缩值;y轴,GO术语。B 强化词网络。每个术语由一个圆形节点表示,其大小与该项下输入基因的数量成正比,颜色代表其簇的身份(即同色节点属于同一簇)。相似度评分为>0.3的项由一条边连接(边的厚度表示相似度评分)。
讨论
本研究引入了基于GNN的新型分类模型,专门设计用于通过分析DTI数据识别SZ个体。我们研究的一个关键方面是解决临床研究领域中验证分类模型在多地点数据集中性能的挑战。这些挑战包括缓解潜在的混杂因素,如扫描设备差异、多样的成像方案以及样本量有限。为有效解决这些问题,我们使用了一个包含来自7个不同研究地点的1507个样本的数据集。为了进一步减少研究地点差异带来的潜在偏差,我们采用ComBat方法消除数据中的具体场地差异。为了增强研究结果的稳健性,我们进行了10折交叉验证实验,使我们能够验证模型在不同子集数据中的表现。此外,我们的模型利用图结构化数据,展示了其捕捉不同脑区间复杂非线性关系的能力,并通过GAT的应用保存关键拓扑信息。与依赖一维特征或使用卷积神经网络(CNN)的二维矩阵运算的传统方法相比,我们的分类模型表现出更优越的性能,凸显了基于GNN方法的优势。我们的结果还强调了SZ与不同脑区之间复杂且非线性的相互作用相关。GAT模型通过使用注意力机制,根据连接的重要性赋予变量权重,有效地捕捉了这些关系。这些非线性关系反映了某些脑区对SZ病理的贡献过高,可能为此前未被识别的疾病进展路径提供见解。然而,由于神经网络本身的复杂性,这些关系仍难以完全解读。我们计划通过整合可解释的人工智能技术,在未来研究中解决这些挑战,以更好地理解模型的决策过程。我们提出的模型在区分SZ患者与头盔患者的分类准确率报告为73.79%。虽然这一准确率超越了偶然性,反映了研究组的平衡性质,但其准确率似乎略低于文献中其他研究的结果。例如,15 使用梯度增强决策树算法实现了约75.05%的准确率,而其他DTI研究中也记录了70%至80%之间的准确率差异。然而,必须认识到可能导致这些性能差异的方法论和数据集差异。由于样品的独特特性,这可能会影响分类结果。此外,特征选择过程也有显著差异;Liang等人从多种神经影像指标中提取特征15,而我们的研究强调的是专门从FA和FN数据中得出的特征。这种有针对性的方法使得对白质连接性进行了更深入的探究,可能影响模型的性能。分类准确率的差异也可归因于不同的评估方法,如交叉验证策略和所采用的算法。最后,这些研究进行的临床环境会影响结果的准确解读。因此,尽管我们的模型表现有竞争力,但在与文献比较时,考虑这些背景因素至关重要。大量研究一致表明,与颅阂患者相比,SZ患者在特定脑区的FA值明显低于头盔。早期研究显示,早期患者在FA评分上明显低于健康患者61。这些FA的变化也延伸到连接精神病患者皮层、小脑、丘脑和皮层的纤维束62。值得注意的是,从左腹侧后扣带皮层到中颞回的通路显示精神疾病患者FA显著减少63。与此同时,研究发现SZ患者大脑连接性有显著变化,包括视觉网络、感觉运动网络、额顶顶网络和顶叶网络的变化64--66。我们的分类模型利用这一有力证据,基于基于特殊FA脑网络和FN脑网络支持的基础数据,适用于SZ患者。应用模型解释技术可以识别对分类任务至关重要的关键脑区。我们的发现揭示了与SZ密切相关的脑区结构变化,包括扣带回、额叶和顶叶。特别是扣带皮层在情绪形成、处理、学习和记忆等过程中起着关键作用。前扣带是显著性网络的中心枢纽,这些区域的变化可能与SZ的注意力缺陷有关67。早期研究显示,与头盔癌相比,SZ患者后扣带回和右颞中回的FA值有所下降68。此外,左肩扣带回和丘脑的FA减少69,与SZ患者扣带束完整性破坏的情况相符70。后续研究表明,工作记忆缺陷------SZ病理生理的一个显著特征------可能与额叶、颞叶和顶叶异常有关71。这些区域与认知功能、语言处理和工作记忆有关。这些区域内网络结构和功能的不规则性可能影响选择性注意力、语言处理和工作记忆执行,可能导致认知功能受损72。研究表明,SZ患者表现出白质异常,特别集中在颞叶(下纵束)和扣带带区73。此外,SZ患者在左外囊、右丘脑-前额叶束和丘脑-顶叶束等多个脑区的FA显著减少74,支持这些区域的结构变化,与我们在额叶和顶叶的发现一致。此外,SZ患者在双侧额叶、右枕上回和左前肢的FA明显较低内囊75,证实这些区域的结构变化,与我们在额叶和顶叶的发现相符。这些区域内网络结构和功能的不规则可能导致认知功能受损,影响选择性注意力、语言处理和工作记忆。这些研究结果加深了我们对SZ患者大脑结构变化的理解,对诊断、治疗和预防具有重要意义。最终,通过富集分析,我们确定了关键基因本体,包括"突触后"、"轴突"和"突触前"。这些结果与此前强调突触病理在SZ中重要性的研究一致76。例如,SZ患者表现出树突状棘密度和谷氨酸能突触后异常77。突触密度的变化被认为与SZ背后的大脑连接性病理变化相关78。单胺轴突的退化和过度生长分别与该病的阴性、认知和阳性症状相关79。突触病理和神经递质系统的异常可能在该病因中起重要作用。因此,理解SZ突触病理的潜在机制对于设计新的治疗策略和干预措施至关重要。本研究所用数据集未收集参与者的遗传数据,也没有针对SZ研究的遗传数据或MRI数据。然而,已有研究利用遗传和MRI数据进行SZ分类,确认了利用遗传数据进行分类的可能性80。将遗传数据整合进我们的分类框架,将显著提升准确性和可解释性。未来研究阶段可探索多模态方法,利用遗传标记和影像特征,丰富我们对SZ神经生物学基础的理解。
总结和局限性
为探讨利用DTI数据对SZ组和HC组患者进行分类的可行性,我们开发了一个基于注意力机制图的模型。我们的研究表明,利用双分支图神经网络模型并从DTI数据中提取多模特征,优于单分支图神经网络模型及其他比较实验方法。基于这种双分支图的注意力机制,我们进一步定位了与分类相关的关键脑区,如上额回、扣带回、中额回、丘脑、眼眶回和前楔形叶。我们还识别出了关键元素,如"突触后"、"轴突"和"突触前"。这些发现与文献一致,有助于理解SZ的神经生物学基础。本研究开发的框架整合了FA和FN脑网络与图谱神经网络,丰富了SZ研究领域,揭示了治疗干预的新途径。通过突出关键的脑区和遗传标志物,我们的工作加深了对单纯性突躁症背后复杂神经机制的理解,并为其诊断、治疗和预防提供了潜在启示。尽管在识别SZ方面取得了显著进展,我们的研究仍面临一些值得关注的挑战。首先,尽管我们使用了精神病学队列中相对较大的样本量,但我们的研究受限于可用的深度学习样本数量。此外,在多个地点进行实验并采用多样化扫描协议,可能影响模型的分类性能。因此,我们建议未来研究在更大群体中采用标准化扫描方案,涵盖来自不同地区的参与者,以增强分类模型的稳健性和普遍性。我们模型的分类准确度可能低于某些报告值,这可归因于本研究所用数据集的多样性------来自多个不同站点、不同协议收集的数据固有地带来了更大的变异性。为此,未来研究可受益于标准化的数据收集和预处理方法,有助于减轻数据异质性带来的噪声。此外,结合其他成像方式并优化模型架构,还能进一步提升分类结果。尽管本研究优先使用FA网络进行基因表达分析,因其在评估白质完整性方面表现出优异性能和既定作用,但我们认识到纳入FN数据可以提供互补的见解。未来的研究有望探讨FA和FN的结合,以更深入理解SZ的遗传基础,从而推动该领域的研究进展。