大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
近日,英国癌症研究所/美国纪念斯隆凯特琳癌症中心Kristian Helin 团队在Nature子刊《Nature Communication 》上发表题为"Addressing the specific roles of histone modifications in transcriptional repression "的科研成果,系统探讨了H3K27me3 、H3K36me3 和H3K9me3三种经典抑制性组蛋白修饰在转录抑制中的重要功能。
研究利用PRC2复合体模块化结构,在H3K27me3缺失的小鼠胚胎干细胞中将H3K36me3或H3K9me3的催化酶定向招募至PRC2靶基因位点并进行分析。发现尽管H3K36me3和H3K9me3两种修饰都能在基因组范围内重建H3K27me3分布模式,但它们均无法完全替代H3K27me3的特异性转录抑制功能, 其抑制效率依赖于特定染色质环境,尤其是启动子区H3K4me3水平。H3K36me3虽能显著降低H3K4me3水平,但残余H3K4me3抑制DNA甲基化有效招募;而H3K9me3具有较强抑制效果,但仍受H3K4me3拮抗。该研究揭示了组蛋白修饰的功能并非简单冗余,而是高度依赖于其所在的特定染色质环境,强调"组蛋白密码"的复杂性和环境依赖性。

****英文标题:****Addressing the specific roles of histone modifications in transcriptional repression
****译文标题:****探究组蛋白修饰在转录抑制中的具体作用
****发表时间:****2025年11月22日
****发表期刊:****Nature Communication
****影响因子:****IF15.7/Q1
****技术平台:****ChIP-seq、RRBS、RNA-seq等
****作者单位:****英国癌症研究所、美国纪念斯隆凯特琳癌症中心、丹麦哥本哈根大学、北京国际癌症研究所
DOI:10.1038/S41467-025-66426-Z
科学家们对表观遗传过程的理解基于一个假说:DNA和组蛋白的单个翻译后修饰或其组合,能够引导产生独特转录下游效应。但组蛋白修饰通常被宽泛地归类为抑制性或激活性,进而引发了关于其潜在功能冗余性的疑问。为此,本研究提出了一种通过将其他组蛋白修饰替代全基因组H3K27me3模式的策略,研究者利用PRC2的模块化结构,在H3K27me3缺失的小鼠胚胎干细胞(mESCs)中,将H3K9me3和H3K36me3定向de novo招募至PRC2靶基因。分析结果显示,尽管在PRC2靶基因上准确重建了全基因组H3K36me3,并导致H3K4me3水平显著降低,但残余H3K4me3抑制了H3K36me3招募足够的DNA甲基化,从而无法替代H3K27me3介导的抑制功能。此外,研究者还证明H3K9me3可更高效抑制H3K27me3调控基因,但这种抑制同样依赖于H3K4me3状态。上述研究结果表明了H3K27me3的特异性抑制功能,并提示单个翻译后修饰的功能效应高度依赖于其与现有染色质环境的互作。
易小结
本研究在基因组范围内直接比较了不同抑制性组蛋白修饰(H3K27me3, H3K9me3, H3K36me3)的功能特异性与可互换性,拓展了将组蛋白修饰简单归类为"激活"或"抑制"的宽泛模式,揭示了表观遗传调控的层次性与环境依赖性。
ChIP-seq和RRBS等高通量测序技术在本研究中发挥了核心作用,前者精准绘制组蛋白修饰和蛋白因子的全基因组分布图谱,后者则定量揭示DNA甲基化动态变化。技术联用为今后类似功能基因组学研究提供了方法学参考,尤其是在需要同时解析多种表观遗传调控的复杂机制研究中。
研究方法
****(1)细胞系构建:****构建Suz12敲除小鼠胚胎干细胞系,通过双gRNA策略诱导大片段缺失;构建RBBP5-FKBP12F36V-2xHA可降解标签细胞系,实现H3K4me3快速缺失;建立稳定表达嵌合融合蛋白(S12N:SD2、S12N:SUV2及其催化失活突变体)细胞系。
****(2)嵌合复合体设计:****基于PRC2模块化结构,将SUZ12的N端招募结构域(S12N,aa 1-544)分别与SETD2的SET催化结构域(H3K36甲基转移酶)或SUV39H2的chromodomain-SET结构域(H3K9甲基转移酶)融合,构建靶向PRC2靶基因的嵌合表观遗传修饰酶。
(3)高通量组学分析
****染色质免疫沉淀测序(ChIP-seq):****在全基因组范围内绘制SUZ12(包括全长、S12N片段及嵌合体)、RBBP5、KDM5B等蛋白的染色质结合谱。这是验证嵌合体靶向准确性和研究蛋白招募动态的关键。
****CUT&RUN:****绘制H3K27me3、H3K4me3、H3K9me3、H3K36me3和H2AK119ub1的全基因组分布图谱,以评估修饰沉积的效率和模式。
****RNA测序(RNA-seq):****全面分析不同基因型细胞中的基因表达变化,定量评估H3K27me3缺失导致的基因去抑制,以及H3K36me3或H3K9me3沉积对基因表达的挽救效果。
****简化基因组亚硫酸盐测序(RRBS):****单碱基分辨率、高深度检测CpG位点的DNA甲基化水平,关注PRC2靶基因区域(CpG岛)的DNA甲基化变化,以探究H3K36me3与DNA甲基化之间的功能关联。
(4)生物化学与细胞功能实验
****体外甲基转移酶活性实验:****使用重组KMT2B复合物和带有或不带H3K36me3修饰的核小体底物,验证H3K36me3对H3K4甲基转移酶活性的直接影响。
****胚胎体分化实验:****评估不同细胞系分化为跳动心肌细胞簇的能力,作为多能性维持和分化潜能的表型读值。
****Western Blot:****检测整体组蛋白修饰水平、蛋白表达和降解效率。
结果图形
(1)构建靶向Polycomb的嵌合表观遗传复合体
本研究首先基于PRC2模块化结构验证用于靶向招募异源酶活性的可行性。研究人员在Suz12敲除(KO)小鼠胚胎干细胞中重新表达SUZ12的N端结构域(S12N,氨基酸1-544),ChIP-seq分析显示S12N能够正确定位于PRC2靶基因区域,其结合模式与全长SUZ12相似,但信号强度有所降低 (图1b)。Western blot证实S12N表达细胞中完全缺失H3K27me3修饰(图1c),且基因表达谱与Suz12 KO细胞高度一致(图1d),表明S12N单独结合不具备转录抑制功能。
基于此,研究团队构建S12N:SD2(融合SETD2的SET结构域,催化H3K36me3)和S12N:SUV2(融合SUV39H2的chromodomain和SET结构域,催化H3K9me3)两种嵌合蛋白。ChIP-seq结果显示,两种嵌合蛋白均能在PRC2靶基因位点实现基因组定位 (图1f)。这一系统成功构建在全基因组范围内将H3K36me3或H3K9me3定向沉积PRC2靶基因的技术平台,为后续功能比较研究奠定基础。
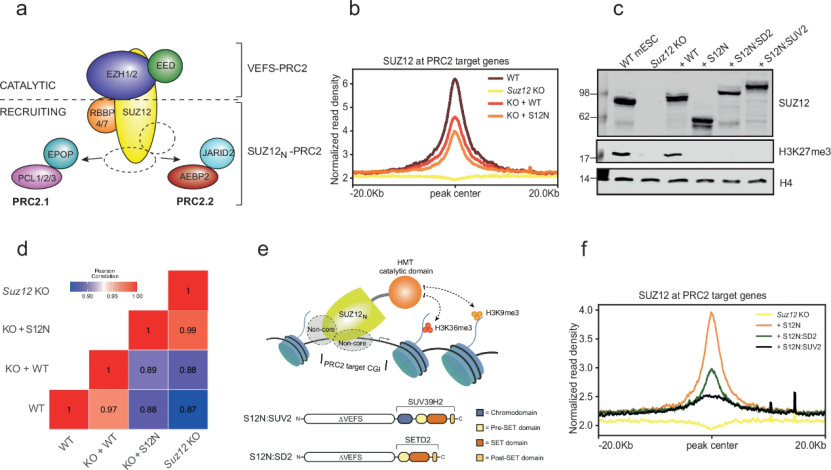
图1:构建靶向Polycomb的嵌合表观遗传复合体
(2)异位H3K36me3沉积在全基因组范围内模拟内源性H3K27me3
为评估S12N:SD2嵌合体沉积H3K36me3的准确性,研究者进行了H3K36me3的CUT&RUN分析。结果显示,在S12N:SD2表达的细胞中,H3K36me3被特异性地沉积在原本富含H3K27me3的区域,例如HoxA基因簇(图2b)。这种沉积模式在基因组内与内源性H3K27me3高度相似(图2f)。通过定量检测所有内源性H3K27me3峰内信号,研究者发现异位H3K36me3的信号强度与野生型细胞中H3K27me3的信号强度显著相关(图2g)。上述CUT&RUN数据证明S12N:SD2系统能够在全基因组范围内以接近生理水平的精度和强度,利用H3K36me3重建H3K27me3分布图谱。
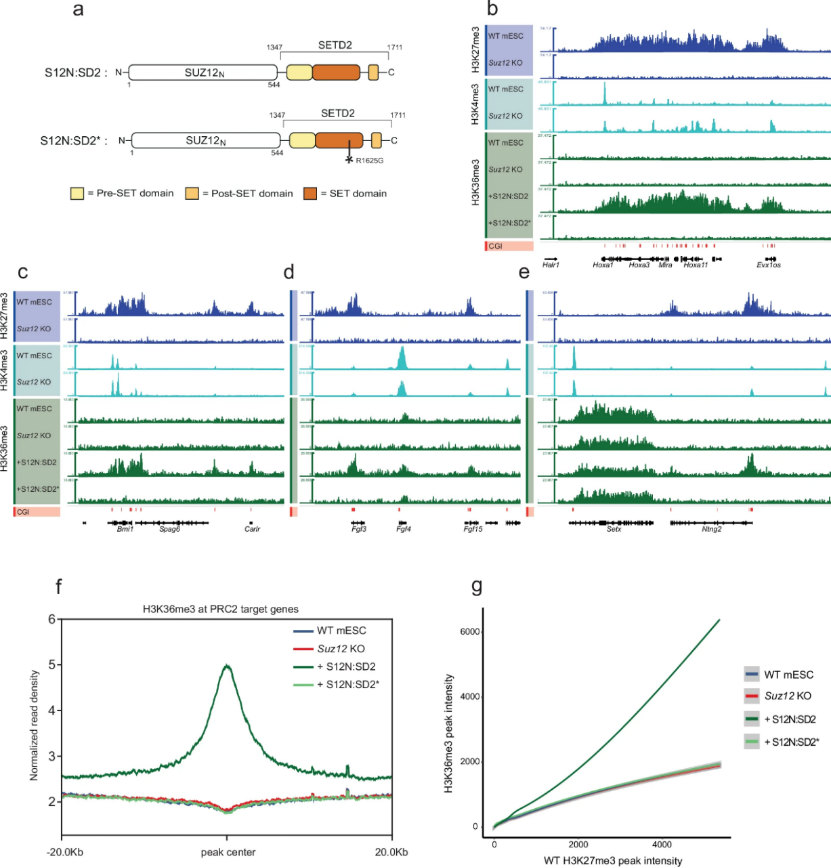
图2:异位H3K36me3沉积在全基因组范围内模拟内源性H3K27me3
(3)H3K36me3无法替代H3K27me3恢复基因抑制和mESC多能性
为评估H3K36me3的功能效应,研究者进行了RNA-seq分析。K-means聚类(K=4)显示,Suz12 KO导致PRC2靶基因广泛去抑制,而野生型SUZ12重建可完全恢复基因表达至野生型水平(图3a)。然而,令人惊讶的是,尽管S12N:SD2在全基因组范围内准确重建了H3K27me3分布模式,却未能普遍抑制PRC2靶基因(图3a)。在SUZ12缺失导致的1326个去抑制PRC2靶基因中,重新表达全长SUZ12(恢复H3K27me3)可以挽救74.1%基因表达,而S12N:SD2(沉积H3K36me3)仅能完全挽救3.5%(图3b-c)。功能实验发现S12N:SD2未能恢复mESC的正常分化能力,类胚体形成实验显示其无法产生跳动的心肌细胞团(图3d),与Suz12 KO细胞类似。
上述研究结果表明,即使一种组蛋白修饰能够在基因组范围准确模拟另一种修饰的空间分布,其功能效应也可能截然不同。H3K36me3通常标记活跃转录基因的基因体区域,其"抑制"功能仅限于抑制隐性转录起始;而H3K27me3则是发育调控基因启动子区的经典抑制标记。这一结果证明组蛋白修饰功能强依赖于其基因组环境,而非化学修饰本身。
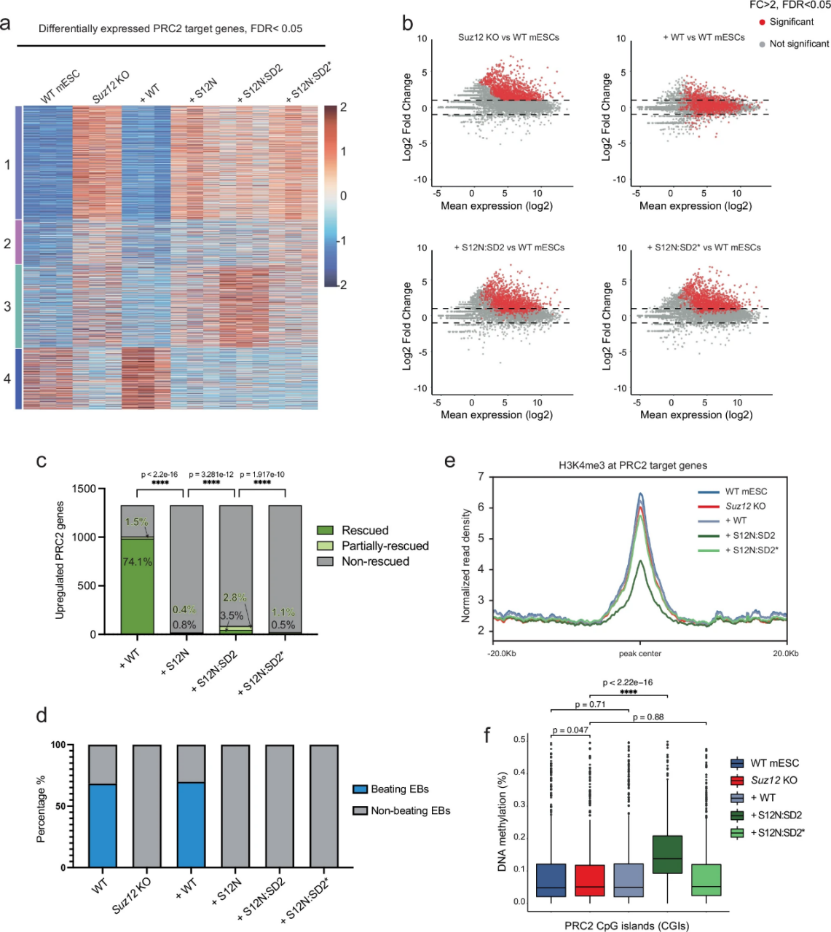
图3:H3K36me3无法替代H3K27me3调控基因表达和分化
(4)Polycomb靶向的H3K36me3显著降低H3K4me3水平
尽管H3K36me3未能有效恢复基因抑制,但其挽救效果(3.5%)仍显著高于S12N单独表达(0.8%)或S12N:SD2*(0.5%),提示H3K36me3在特定染色质环境中可能具有弱抑制活性。为探索这一选择性挽救机制,研究者分析了H3K36me3沉积对染色质环境的直接作用。意外发现,S12N:SD2驱动的H3K36me3沉积导致PRC2靶基因启动子区H3K4me3水平显著降低,且在被H3K36me3挽救的基因中这一降低更为显著(图3e)。进一步研究表明,启动子区H3K4me3降低主要源于H3K36me3对H3K4甲基转移酶催化效率的直接抑制,而非复合体招募(RBBP5)或去甲基化酶(KDM5B)活性改变。这一发现揭示了组蛋白修饰之间的直接互作,H3K36me3可通过空间位阻或变构效应抑制H3K4甲基化,为理解修饰间层级关系提供了新视角。
(5)残余H3K4me3通过拮抗de novo DNA甲基化阻碍H3K36me3介导的抑制
先前已有研究将H3K36me3介导的基因抑制与DNMT3B招募和de novo DNA甲基化联系起来,这一机制通常与转录延伸偶联,防止基因体内隐性转录起始。值得注意的是,这些区域通常缺乏H3K4me3,而H3K4me3已知可保护CpG岛(CGI)启动子免受de novo DNA甲基化。基于这一背景,研究人员假设尽管H3K36me3在启动子区占据优势,但残余的H3K4me3可能保护CGI免受H3K36me3招募的DNA甲基化,从而阻碍完全抑制。为验证此假设,研究人员进行了RRBS分析,聚焦于野生型H3K27me3峰内的差异甲基化CpG(DMCs)。差异甲基化区域(DMRs)分析显示S12N:SD2驱动的启动子区H3K36me3沉积确实导致了CGI的de novo DNA甲基化 (图3f),证实了H3K36me3与DNA甲基化的联系。
然而,这种甲基化水平在细胞群体中可能不足以引发沉默。更重要的是,在那些被H3K36me3部分挽救的基因中,其初始H3K4me3水平更低,而DNA甲基化水平则更高。研究者****在RBBP5可降解的细胞背景中结合S12N:SD2表达并去除H3K4me3后,RRBS和RNA-seq数据共同显示,DNA甲基化水平进一步升高,同时被H3K36me3抑制的基因数量显著增加。****这证明,残留的H3K4me3是抑制H3K36me3通过DNA甲基化通路实现有效抑制的关键因子。
(6)Polycomb靶向的H3K9me3呈现连续块状积累,但与内源性H3K27me3有差异
为比较H3K9me3与H3K27me3的功能,研究者在Suz12 KO mESCs中表达S12N:SUV2及其催化失活突变体S12N:SUV2*(SUV39H2的F259A和Y261A活性位点突变)(图4a)。分析结果显示与H3K36me3类似,S12N:SUV2表达导致PRC2靶基因位点沉积H3K9me3,但其分布模式与野生型H3K27me3存在显著差异(图4b)。H3K9me3的峰经常延展至H3K27me3区域之外,且其积累并非连续的"块状",而是在CpG岛和H3K4me3富集的位置出现"凹陷"(图4c-e),表明H3K4me3可能拮抗H3K9me3沉积。尽管如此,全局水平上的异位H3K9me3峰强度与内源性H3K27me3信号仍存在相关性(图4f-g)。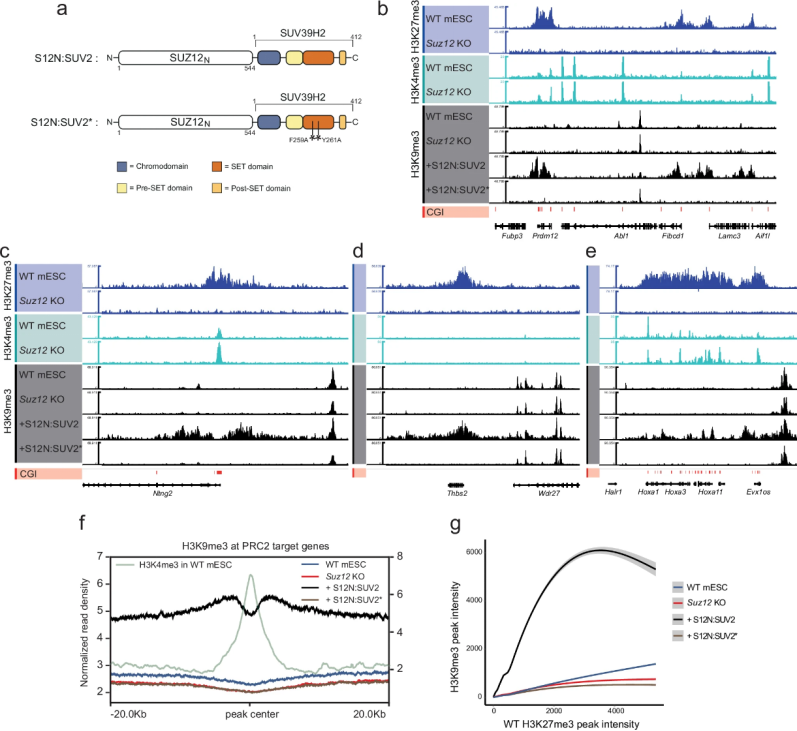
图4:H3K9me3在内源性Polycomb靶基因上的沉积
(7)H3K9me3能以环境依赖方式抑制Polycomb靶基因,直接依赖于H3K4me3状态
与H3K36me3相比,H3K9me3表现出更强的抑制能力。RNA-seq分析表明,S12N:SUV2能完全挽救25.4%的去抑制PRC2靶基因(图5a-c)。然而,其抑制效果同样不完整,且无法恢复正常的细胞分化(图5d)。关键区别在于,H3K9me3沉积并未介导H3K4me3水平降低(图5e),但其抑制效率与靶点原有H3K4me3水平呈强负相关。那些被H3K9me3成功挽救的基因,其启动子在野生型状态下就具有显著较低的H3K4me3水平,从而允许H3K9me3更有效地积累(图5f-g)。当在可降解H3K4me3的细胞背景中表达S12N:SUV2并去除H3K4me3后,ChIP-seq/CUT&RUN显示S12N:SUV2的染色质结合和H3K9me3沉积大幅增强,RNA-seq证实被挽救基因比例提升82.4%(图5h-i)。上述研究结果表明,H3K4me3通过抑制H3K9me3甲基转移酶复合体的稳定染色质结合来直接拮抗H3K9me3沉积。
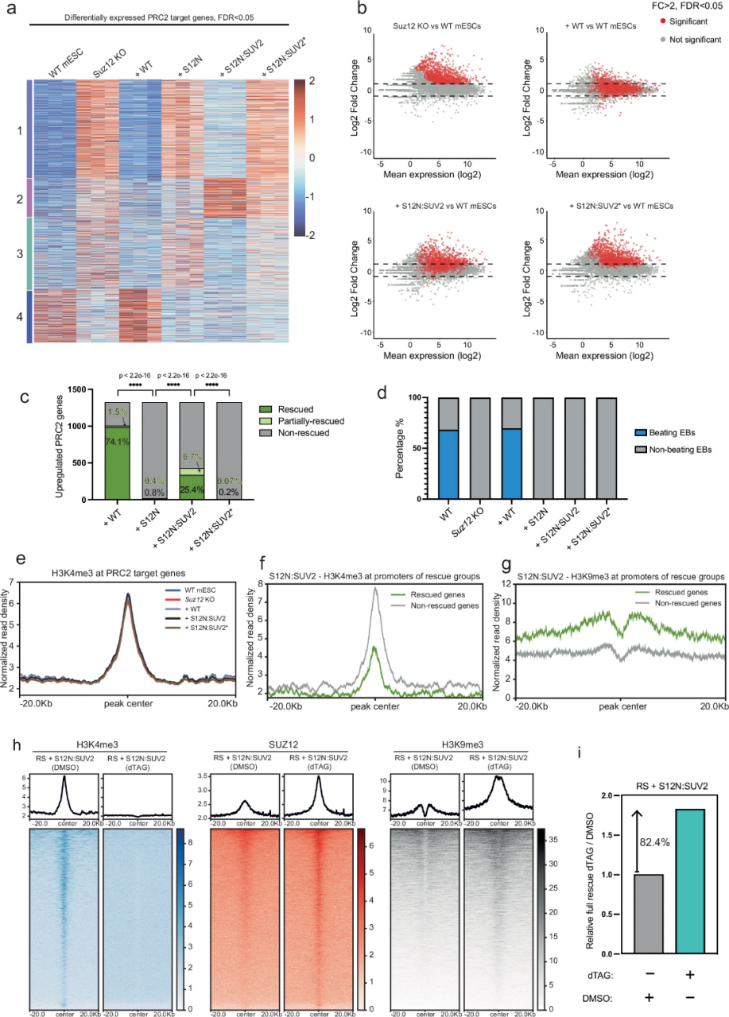
图5:H3K9me3能够以环境依赖方式抑制Polycomb靶基因,取决于其H3K4me3状态
结论和启示
本研究通过ChIP-seq/CUT&RUN+RNA-seq+RRBS多组学整合分析,揭示了H3K9me3、H3K27me3和H3K36me3三种经典抑制性组蛋白修饰的功能并非可替代和互换。H3K27me3在发育调控基因启动子区的特异性功能无法被其他修饰替代,其功能实现强依赖于特定染色质环境,特别是与H3K4me3的互作。H3K4me3通过拮抗H3K9me3和抑制DNA甲基化,保护CpG岛启动子,进而揭示H3K27me3在Polycomb靶基因抑制中不可替代的核心地位,同时阐明了H3K4me3作为染色质"守门人"的关键功能。
ChIP-seq和RRBS在本研究中的重要作用
本研究中的ChIP-seq不仅用于验证嵌合蛋白的靶向准确性(图1b、f),更创新性解析了H3K9me3的异位沉积模式(图4),并通过时程分析揭示了H3K4me3对复合体招募的动态功能(图5h),助力研究者构建"修饰沉积-蛋白结合-基因表达"的关联网络。
RRBS实现了启动子区DNA甲基化的单碱基分辨率定量检测(图3f),揭示了H3K36me3介导抑制失败的关键机制:H3K4me3对de novo DNA甲基化的保护作用。
两种技术联合应用能够在基因组范围内同时追踪组蛋白修饰和DNA甲基化的动态变化,解析之间的功能互作,从定位、功能探索到下游机制,层层深入地解析表观遗传网络。
参考文献:Hedehus L, Mas AM, Alpsoy A, Armstrong RL, Koche R, Tatar T, Wang H, Helin K. Addressing the specific roles of histone modifications in transcriptional repression. Nat Commun. 2025 Nov 22. doi: 10.1038/s41467-025-66426-z.