
1. 小编导读
对于关注复杂疾病遗传学、群体遗传学及多基因风险评分应用的生信研究者而言,这篇于2026年4月正式发表于《自然·遗传学》的研究可谓意义非凡。它不仅是目前规模最大的近视跨祖先全基因组关联研究(样本量超过176万),更在方法学上展示了如何通过大规模数据整合、跨祖先分析、功能注释整合及精细定位,系统性地解析一个复杂性状的遗传架构,并推动其向临床预测应用转化。
2. 摘要
屈光不正(近视、远视、散光)影响着全球超过一半的人口,可导致视力模糊甚至失明。本研究对来自欧洲(149.5万人)、东亚(12.1万人)和非洲(14.5万人)血统的人群分别进行了全基因组关联研究荟萃分析,并开展了跨祖先荟萃分析。跨祖先分析共鉴定出932个与屈光不正相关的遗传变异,包括241个此前未知的关联,以及4个东亚特异性、1个非洲特异性关联信号。通过统计精细定位,研究锁定了16个高可信度的潜在因果变异。基因优先级分析则突出了23个参与眼球发育的基因。研究者构建了一个整合功能注释的增强型多基因预测器,可解释21.4%的屈光不正表型变异,能有效对近视的发病、进展和严重程度进行风险分层,并在预测高度近视时达到了0.806的曲线下面积。此项多祖先GWAS研究极大地扩充了屈光不正的遗传变异图谱,并展示了多基因风险评分在跨种族人群中识别高风险个体的临床潜力。
3. 研究方法
本研究的分析流程和数据规模极具代表性。
样本与表型。 欧洲人群的核心数据来自英国生物银行,包含10.7万人的直接测量等效球镜。此外,研究团队利用一个多项式模型(纳入性别、年龄、出生年份及戴镜起始年龄),为另外30.1万仅有戴镜年龄信息的UKB参与者推算了MSE值。其他补充队列包括23andMe、芬兰基因组计划、百万退伍军人计划、全民健康研究计划等,提供了大量基于ICD编码或自述的近视与对照状态。东亚与非洲人群的数据来源包括日本东北大学医学巨库组织、WeGene、23Mofang、MVP及AllofUs等队列,其中近视与对照表型为主要分析对象。研究还使用EPIC‑Norfolk、ALSPAC以及UKB中的非欧洲裔样本进行独立验证。
统计分析与GWAS荟萃分析。 单祖先荟萃分析采用样本量加权固定效应模型。需要特别指出,作者最初尝试过MTAG方法,但在审稿过程中发现当合并的队列异质性较大时,MTAG可能导致统计量偏差,因此最终选择了更为稳健的传统方法,并对存在潜在膨胀(LDSC回归截距>0.1)的队列进行了统计量校正。跨祖先荟萃分析同样采用样本量加权固定效应模型,以发现不同人群中共享的遗传信号。对于遗传相关性的估计,研究使用Popcorn软件,重点关注其"遗传影响相关性"输出结果,该方法能有效修正样本量差异带来的影响。
遗传力分解。 使用GCTA软件的GREML‑LDMS方法,基于95,519名无亲缘关系的UKB欧裔个体,分解了不同等位基因频率和连锁不平衡分层的SNP遗传力。该方法发现低频与罕见变异(MAF<1%)和低LD区域的变异对遗传力的贡献超过中性进化模型的预期,提示负选择压力的存在。
基因优先级与功能注释。 研究综合运用了十种方法交叉验证以锁定最可能的相关基因,包括基因水平关联检验(MAGMA和mBAT‑combo)、共定位分析(使用COLOC将GWAS信号与eQTL、sQTL、pQTL信号进行共定位,以PP4>0.8为阈值)、转录组与蛋白质组关联研究(使用SMR和FUSION,整合TWAS,纳入GTEx、BrainMeta、INTERVAL等多个参考数据集的分子量)以及功能相似性方法(使用PoPS,基于基因功能相似性进行排序)。对至少被5种方法优先级排序的498个基因进行GO富集分析,发现"眼球发育"等生物学过程显著富集。
跨祖先精细定位。 在单祖先精细定位中,使用CARMA方法整合GWAS荟萃分析汇总数据和功能注释,计算每个变异的后验包含概率。研究者还开发了一个优先级评分系统,计算每个变异在六种不同的精细定位策略(例如CARMA有无功能注释、PolyFun+SuSiE/FINEMAP等)中PIP大于0.5的次数,得分越高的变异其因果可能性越大。在跨祖先精细定位中,使用SuSiEx直接利用多个祖先群体的GWAS汇总数据和LD参考面板,识别跨种族共享的因果变异可信集。
多基因风险评分构建与验证。 采用十折交叉验证策略在UKB欧裔人群中评估PRS性能。每折中,训练集用于重新进行GWAS荟萃分析并估计效应值,测试集用于计算PRS与表型的相关性。PRS生成方法使用SBayesRC,该方法在SBayesR(假设SNP效应服从混合正态分布)的基础上进一步整合了功能基因组注释,显著提高了预测精度。对于跨祖先PRS,使用SBayesRC‑multi方法:先分别为每个祖先训练一个SBayesRC模型,再在调优样本集中学习最优权重进行整合,从而提升了PRS对非欧洲裔人群的预测能力。
4. 主要结果
遗传力与遗传架构。 GREML‑LDMS分析显示,屈光不正的SNP遗传力为0.460(标准误0.013),其中常见变异贡献了0.395,而低频与罕见变异贡献了0.045。低连锁不平衡区域的变异解释了更多的遗传力,这是负选择的特征。屈光不正的遗传架构在三个人群中高度相似:遗传相关性分别为欧‑东亚0.80、欧‑非洲0.88、东亚‑非洲0.79。用欧裔GWAS鉴定的位点(仅占所有SNP的8.8%)能够解释东亚裔81.8%和非洲裔58.2%的SNP遗传力,再次印证了共享的遗传基础。
新位点发现与祖先特异性信号。 跨祖先荟萃分析鉴定出932个准独立关联变异,分布在575个非重叠基因组区域,其中241个是首次报道。东亚特异性信号例子是PDE4B基因座的rs12063046,该位点在东亚人群中频率较高(MAF=0.27),在欧洲却很低(MAF=0.03),此前研究已证实PDE4b基因敲除小鼠会出现明显的近视。非洲特异性信号例子为rs1646026。此外,许多新发现的变异显示出多效性,例如与身高、教育程度、血细胞计数等表型相关联,提示近视与这些复杂性状间存在共同的遗传基础。
基因优先级与生物学机制。 通过多方法整合,研究确定了23个与"眼球发育"生物学过程明确相关的基因,为后续功能实验提供了高质量靶点。这些基因包括SIX3(早期眼发育转录因子)、PRSS56(后部微小眼球相关)、FBN1(马凡综合征相关,眼球轴长增加)等。
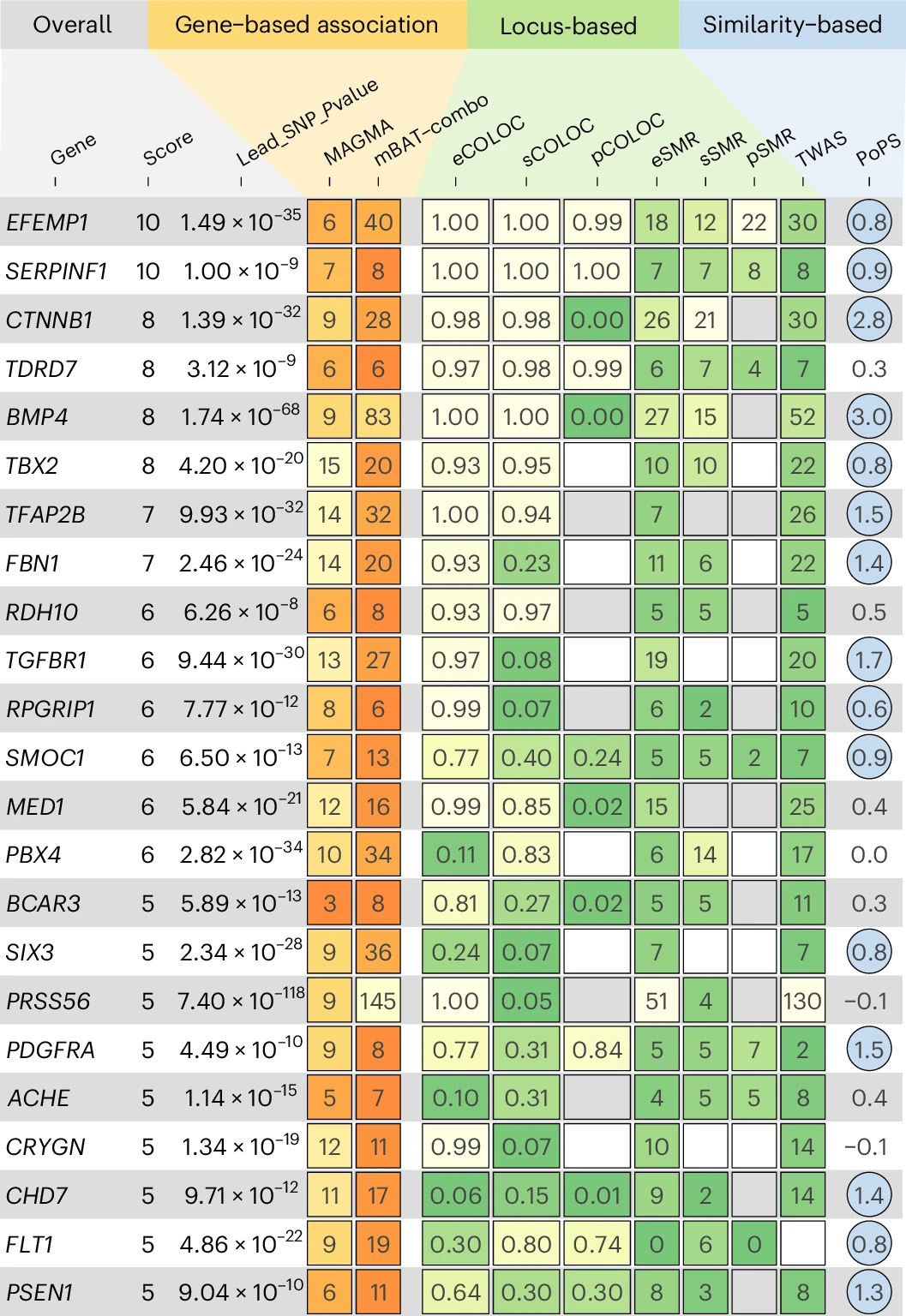
精细定位因果变异。 通过六种精细定位策略的"优先评分系统",有16个变异在全部六种方法中均获得高置信度(PIP>0.5)。这些高可信变异包括位于GJD2基因座的rs634990和位于APOE基因座的rs429358。虽然它们被精细定位为最可能的因果变异,作者谨慎指出其真正的因果角色仍需实验验证。
多基因风险评分的预测性能。 在十折交叉验证中,整合了功能注释的SBayesRC PRS在欧裔人群中可解释21.4%的MSE表型变异,达到了SNP遗传力的54.2%。对近视(MSE ≤ -0.75D)的预测AUC为0.741,对高度近视(MSE ≤ -6.00D)的预测AUC为0.806。在风险分层能力方面,PRS最低的1%人群(近视高风险)中84%为近视,35%为高度近视;而PRS最高的1%人群中仅3%为近视,两者患病率相差28倍。在发病年龄分层方面,PRS越低(近视风险越高)的人群,其戴镜起始年龄的中位数显著更小:最低PRS十分位人群为15岁,最高PRS十分位人群为42岁。在跨种族预测方面,使用欧裔GWAS训练的PRS在东亚裔中预测效率显著下降(R²=8.8%),而采用跨祖先PRS(SBayesRC‑multi)后,在东亚裔中的预测R²提升至9.7%,相比已有最佳PRS提高了115.6%,证明了跨种族预测策略的重要价值。
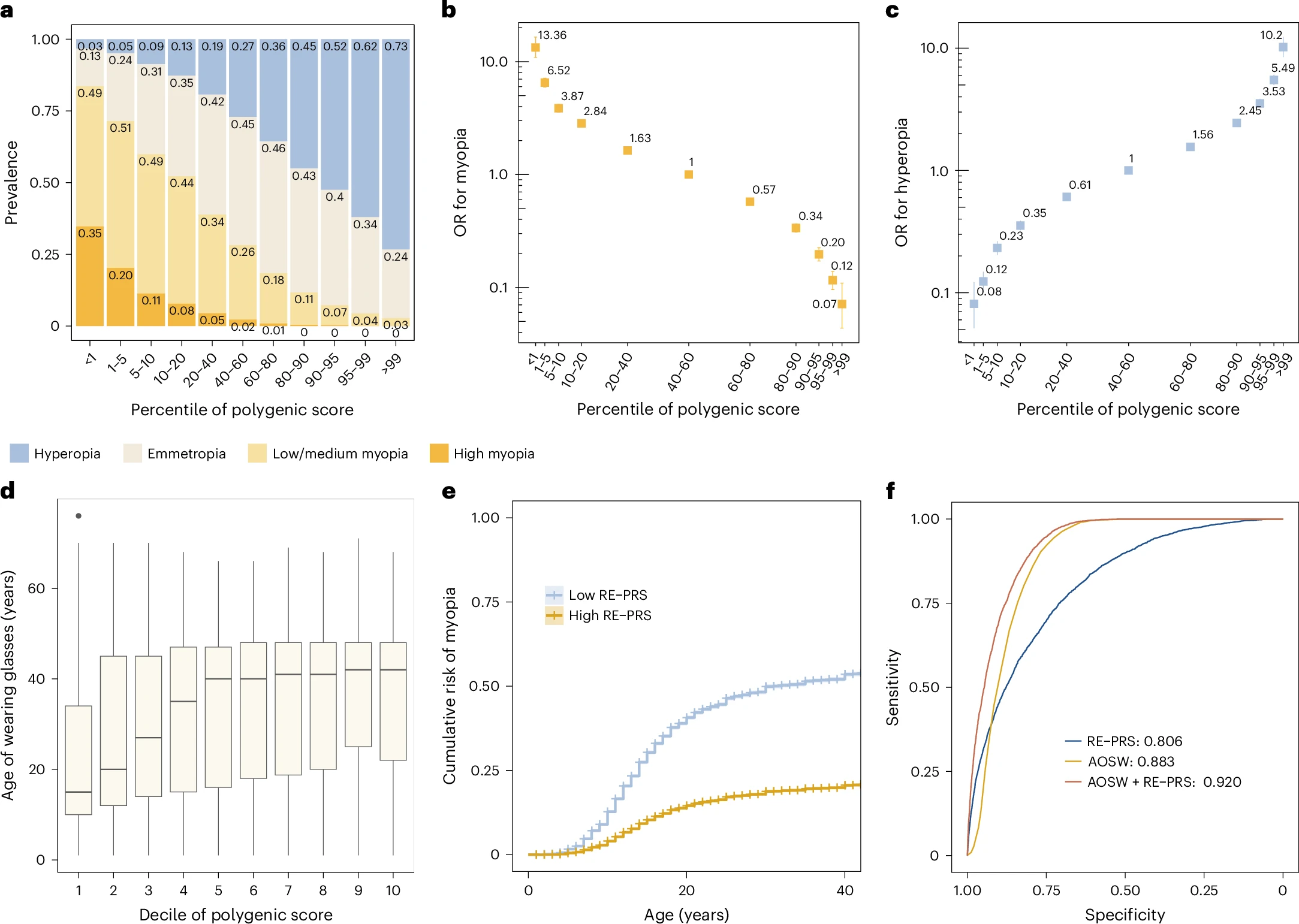
临床联合预测。 尽管AOSW是预测高度近视的强指标(AUC=0.883),但当将PRS与AOSW联合使用时,预测高度近视的AUC可进一步提升至0.920(Delong检验,P=6.1×10⁻¹⁴),显示出PRS在出生时即可提供独立于AOSW的额外预测信息,对早期干预具有重大意义。
5. 结论
本研究通过迄今为止最大规模、最多元祖先的GWAS荟萃分析,极大地丰富了人们对屈光不正遗传架构的理解。研究不仅发现了大量新的遗传位点和祖先特异性信号,还通过系统的精细定位和基因优先级分析,为揭示近视的发病机制提供了高质量的假说驱动靶点。更重要的是,通过整合功能注释和多祖先数据,研究开发的增强型多基因风险评分在风险分层和跨种族预测方面取得了显著进展,展示了将遗传发现转化为精准预防策略的巨大潜力。尽管仍有局限性,例如非欧裔样本量仍相对不足,但该研究为全基因组关联研究如何走向临床实践提供了一个范例。
6. 参考文献
Cheng, FF., Liu, X., Mi, H. et al. Multi‑ancestry genome‑wide association analyses of refractive error augment genetic discovery and polygenic prediction. Nat Genet (2026). https://doi.org/10.1038/s41588-026-02576-0