🖊小编导读
美洲是人类扩散史上最后一块大陆,但美洲原住民的基因组研究在全球主要人群中恰恰是最薄弱的一环。长期以来,SNP芯片数据的内在偏差、样本覆盖的地理局限,以及对 Suruí、Karitiana 等少数人群的过度使用,使得许多关键问题悬而未决。这篇文章发布了迄今规模最大的美洲原住民高覆盖度全基因组数据集(128 个个体,~44×),横跨 8 个拉丁美洲国家、45 个人群、28 个语系,并系统整合了古代 DNA 数据展开分析。对于做人类群体基因组、演化或医学相关研究的同学,这篇文章无论在数据资源还是方法参考上都很有价值。

摘要
背景
美洲原住民代表着人类全球扩散的最后一次主要浪潮,然而在现有基因组数据库中,他们却是主要大陆人群中研究最为匮乏的群体。由于高质量全基因组数据的严重不足,围绕这一群体的演化历史、自然选择信号以及古人类混入等核心问题至今仍未厘清。
结果
本研究发布了 128 个高覆盖度美洲原住民基因组,揭示了大量此前未被描述的遗传多样性。数据显示,南美洲经历了至少三次独立的人群扩散入迁,之后在各地区形成区域分化并长期保持遗传连续性。全基因组自然选择扫描识别出与免疫、代谢、生殖和发育相关基因上的广泛选择信号。此外,多个基因组区域与澳大利亚-美拉尼西亚(Australasian)人群存在显著的等位基因共享,其来源可能是一次古老的混合事件,部分信号在自然选择的维持下延续了超过一万年。研究还检测到来自古人类(尼安德特人和丹尼索瓦人)的适应性渗入。值得注意的是,澳洲血缘相关区域与古人类渗入区域几乎不重叠,说明两类信号的演化起源是相互独立的。
结论
这些发现挑战了以往对美洲大陆定居过程的简单化模型,揭示了美洲原住民更为动态、复杂的演化历史。
方法概览
本研究涉及的生信工具和参数相当密集,以下按分析模块整理:
| 分析模块 | 工具/方法 | 关键参数与说明 |
|---|---|---|
| 测序与数据质控 | 全基因组测序(WGS) | 平均覆盖度 ~44×,共 128 个新测序个体 |
| 群体结构 | PCA | 基于 LD pruned、masked 的全基因组 SNP,限制无亲缘关系个体(160人) |
| 祖先比例推断 | ADMIXTURE(非监督模式) | 最优 K 值:整体 K=12,古今联合分析 K=10;结果可视化使用 PONG |
| 遗传距离与聚类 | outgroup f3 统计量 + Neighbour-joining tree | 以 Mbuti 为外群,计算 1 − f3(Mbuti; X, Y);NJ tree 可规避群体特异漂移带来的偏差 |
| 迁移模式 | EEMS(Estimated Effective Migration Surface) | 模型下偏离隔离距离(IBD)的基因流差异可视化 |
| 近亲系数与纯合片段 | ROH 分析 + F_ROH | 利用纯合连续片段(ROH)计算个体近交系数,区分古老瓶颈与近代近交 |
| 血缘同一性 | IBD 分析 | 分析 >8 cM 片段的群体内外共享比例,推断近期基因流与有效种群大小 (Ne) |
| 有效种群大小 | IBD-based Ne + 群体内溯祖率(coalescence rate) | 两种方法互补:IBD 捕捉近 50--100 代事件;溯祖方法分辨古老历史 |
| 种群分化时间 | Cross-coalescence rate | 估算不同遗传簇与其他大陆人群的分化时间 |
| 混合图建模 | admixtools R 包(qpgraph、qpAdm、qpWave、find_graphs) | find_graphs 自动搜索最优混合图,迭代添加 0--5 次混合事件;qpAdm 推断祖先来源比例;qpWave 检验祖先流数目;所有分析仅使用 transversions 以减少偏差;置信区间通过 SNP-block resampling 计算 |
| 自然选择扫描 | PBS(Population Branch Statistic)+ EHH(Extended Haplotype Homozygosity) | 基于群体分化与单倍型延伸联合打分(Fisher combined score),滑动窗口大小 200 SNPs、步长 50 SNPs |
| 澳洲血缘检测 | D-statistics(ABBA-BABA) | D(Mbuti, Onge; Mixe, X) 检测超额澳洲亲缘性;与尼安德特人/丹尼索瓦人亲缘性独立对照 |
| 祖先遮蔽 | Local ancestry inference + masking | 针对存在欧洲/非洲混入的个体,推断本地祖先并遮蔽非原住民片段 |
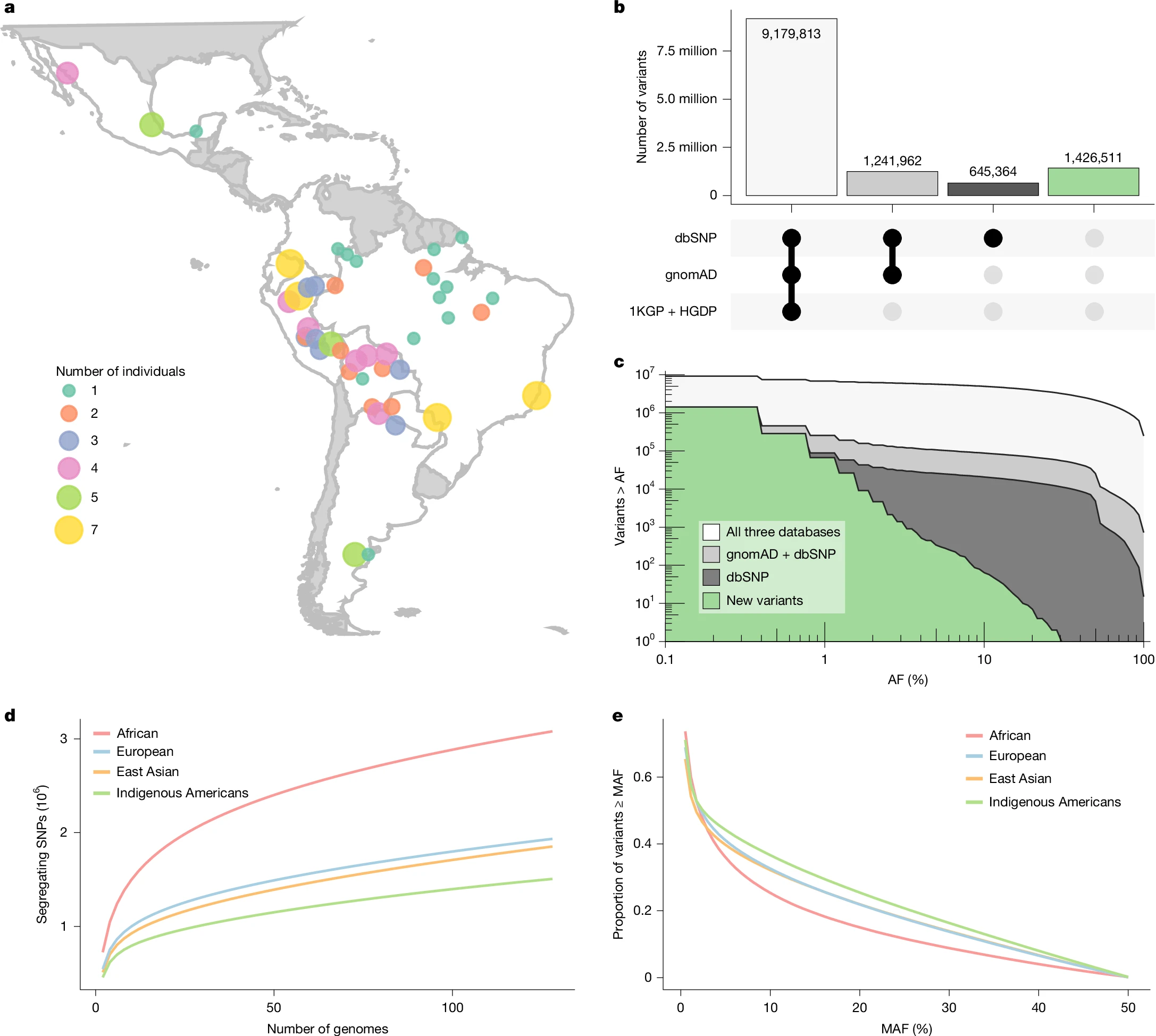
| 新变异识别 | 与公共数据库比较(1KGP + HGDP、gnomAD、dbSNP) | 逐步过滤(UpSet plot),总 SNV 12,493,650;新变异 1,426,511(11.4%) |
主要结果
一、美洲原住民拥有大量未被收录的遗传多样性
将 128 个高覆盖度基因组与超过 27 万人的公共数据库(1KGP、HGDP、SGDP、gnomAD、dbSNP)比对后,研究共识别出 12,493,650 个 SNV,其中 11.4%(约 142.6 万个)为数据库中未收录的新变异。每个个体平均携带约 11,100 个新变异,虽然低于非洲人群(约 27,800 个),但与大洋洲人群(约 12,600 个)相当。这说明:美洲原住民虽整体多样性相对偏低,但仍存在大量此前被主流数据库遗漏的独特变异,也进一步凸显了将这一人群纳入基因组研究的紧迫性。
二、群体结构与地理格局高度吻合
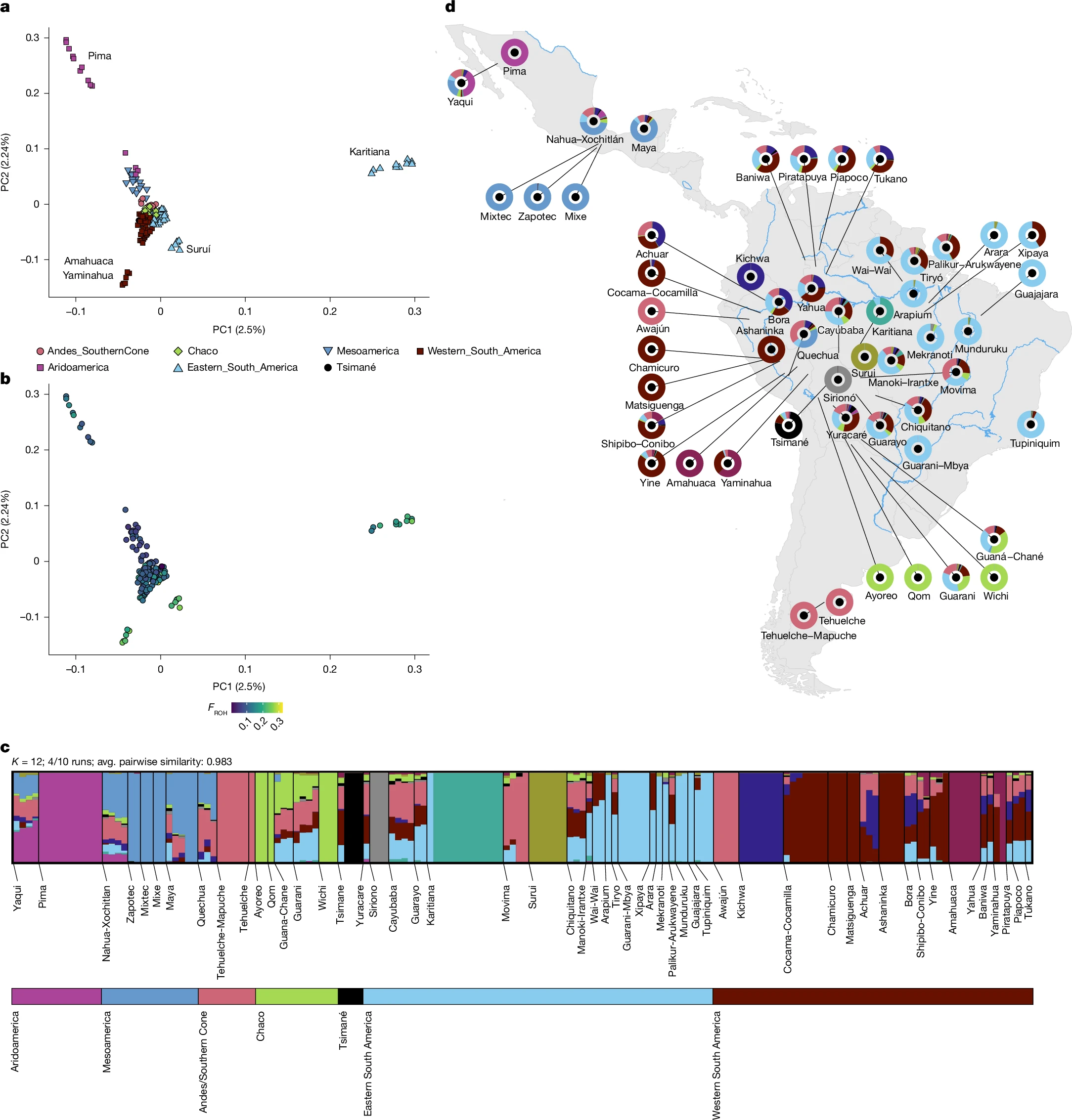
基于 PCA 和 ADMIXTURE(K=12)的分析,北美与南美人群之间存在清晰分化,南美内部进一步分为南锥(Southern Cone)、东南美、西南美低地和查科(Chaco)四个主要遗传簇。需要特别指出的是,Karitiana 和 Suruí 这两个长期被用作南美低地遗传代理的人群,由于近亲系数(F_ROH)显著偏高,其遗传分化程度在同地区人群中也是最大的,并不适合作为低地南美遗传多样性的通用代表。北美方面,Pima 和 Yaqui 最先从其他人群中分化出来,其后依次是 Nahua、Mixe/Mixtec/Zapotec 及 Maya,与考古学上 Aridoamerica 和 Mesoamerica 的划分吻合。
此外,分子方差分析(AMOVA)显示遗传簇能解释约 9% 的变异(P=0.04),而民族语言分组则没有显著解释力(P=0.57)。EEMS 分析进一步显示,秘鲁东南、玻利维亚北部和亚马逊西南是遗传分化超出地理距离预期的热点区域,Mesoamerica 与南美之间则存在高于预期的基因流。
三、殖民历史在基因组上留下的痕迹
ROH 分析显示,Moseten(Tsimané)、Pano-Takana(Amahuaca、Yaminahua)、Zamuco(Ayoreo)和 Tupi(Sirionó、Suruí、Karitiana)语系人群拥有最高的 ROH 数量和总长度,且存在大量长片段 ROH,这很可能是欧洲殖民导致的人口崩溃、隔离与碎片化的直接遗传痕迹。基于 IBD 的 N e 推断也显示了殖民接触后各人群普遍存在的瓶颈效应,仅 Chaco 和西南美部分地区出现了近期的种群恢复迹象。
四、南美至少经历三次独立的人群扩散
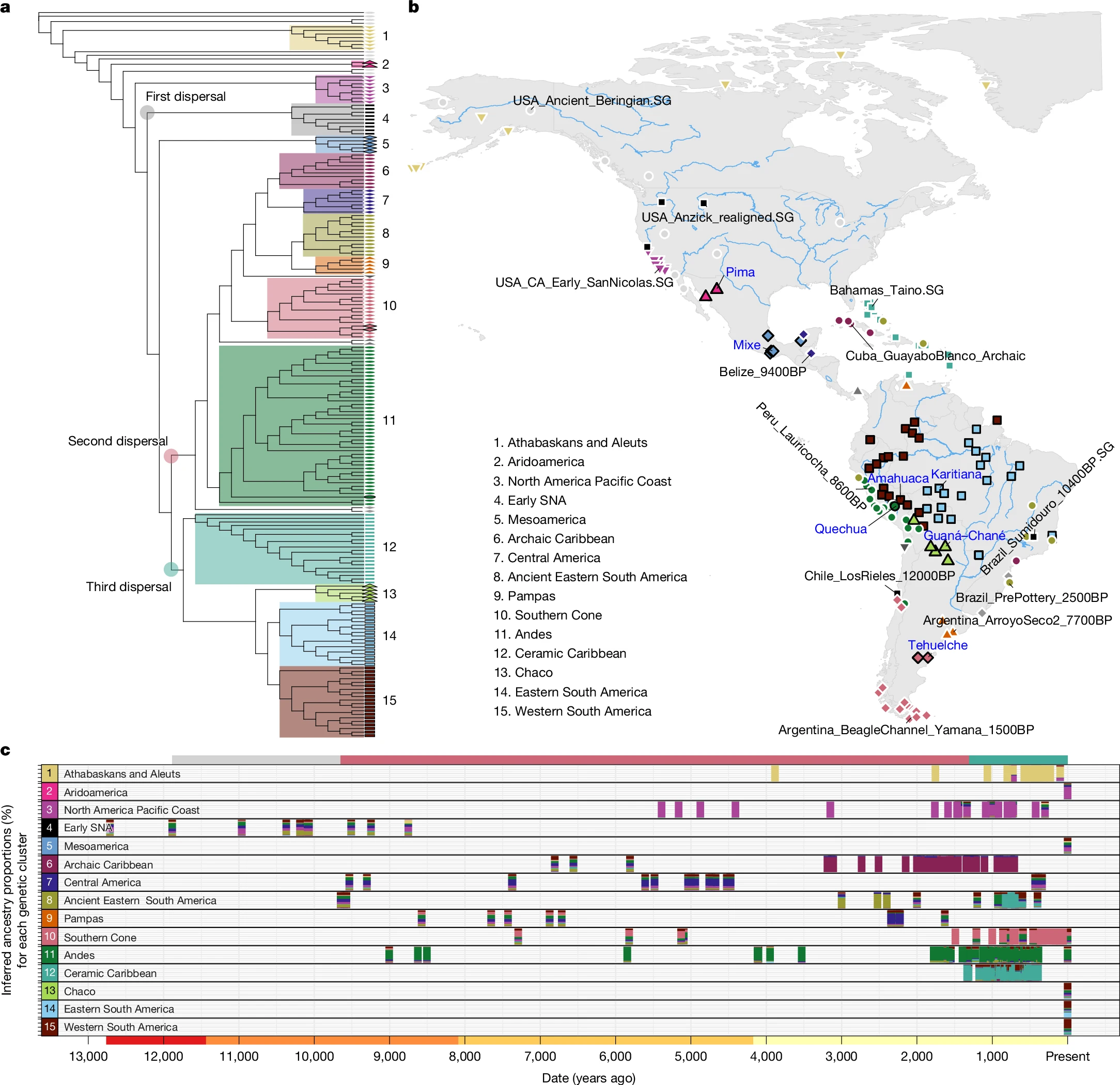
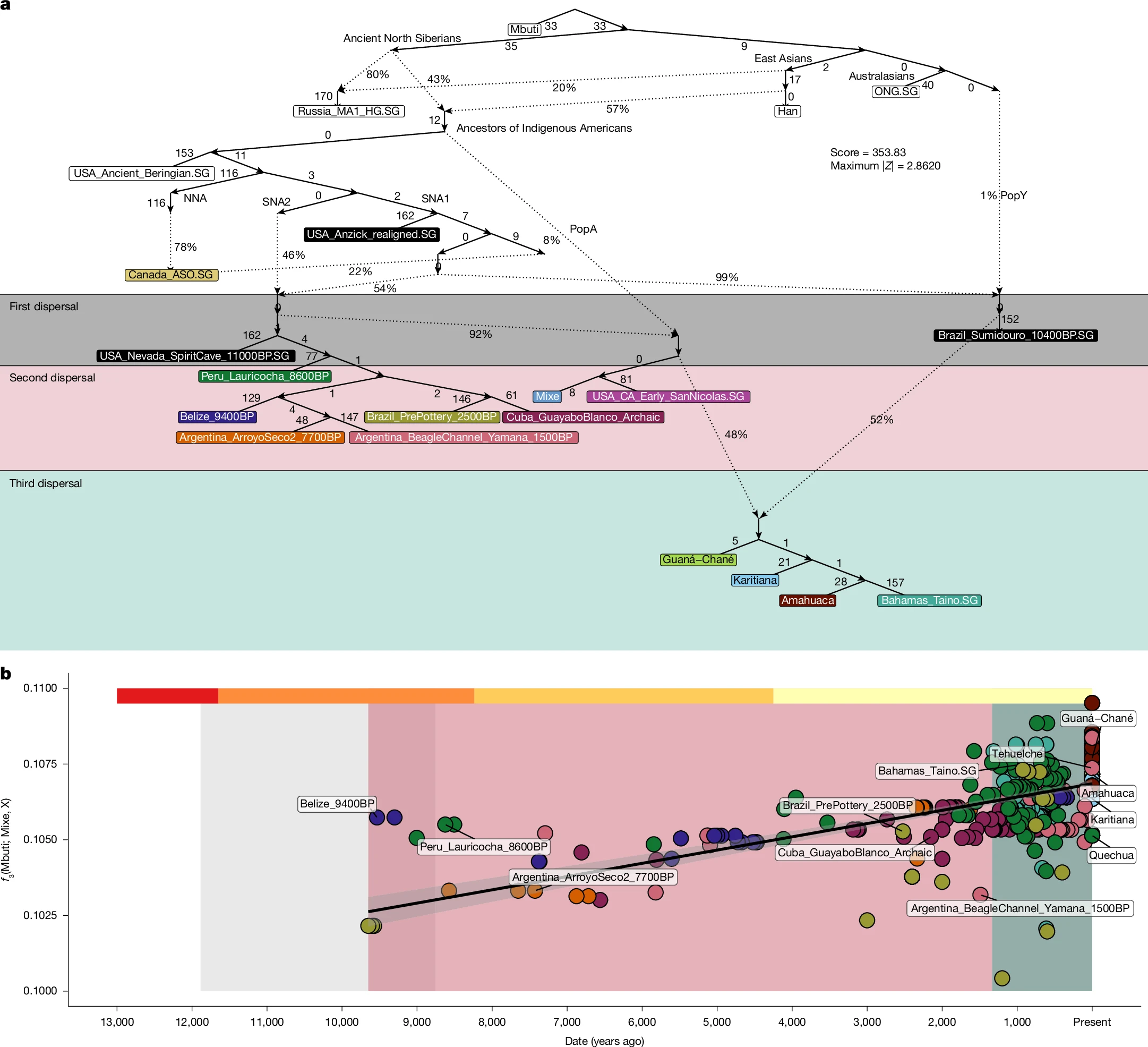
整合现代和古代基因组(含 Allen Ancient DNA Resource 数据库及巴西 sambaqui 古人)后,研究构建的 NJ 树揭示了南美洲至少三次主要的入迁事件。第一次扩散发生在 9,000 年前以前,代表人物包括 Anzick-1(约 12,700 年前,Clovis 文化)、Spirit Cave(约 11,000 年前)、Lagoa Santa/Sumidouro 等早期南美本地人(SNA);第二次约 9,000 年前从中美洲向南扩散,留下了 Archaic 期加勒比人、部分安第斯和南锥人群的遗传连续性;第三次则是此前未被识别的一次晚期扩散,覆盖了几乎所有现代南美原住民和陶器时代(约 2,500--500 年前)的加勒比人,来源可能与中美洲(Mesoamerica)相关人群有关,最早发生于距今至少 1,300 年前。qpWave 检验排除了用 1--2 个祖先来源解释第三次扩散多样性的可能(P=0 和 P=1.8×10⁻²⁵),至少需要三个遗传来源。
五、与澳大利亚-美拉尼西亚人群的古老遗传亲缘
部分美洲原住民(Awajún、Ayoreo、Guarani、Karitiana、Sirionó、Suruí、Tsimané 等)相对于同大陆其他人群,对澳大利亚-美拉尼西亚人群(Australasians)表现出显著的超额遗传亲缘性(Z>3),在亚马逊西南部最为集中。这一信号最早出现在约 10,400 年前的 Sumidouro 古人身上,并延续至全新世晚期。D 统计量分析证实,这种澳洲亲缘性与尼安德特人和丹尼索瓦人渗入之间没有相关性(r=-0.006 和 r=-0.10),说明两者是独立来源的信号,而非共同的古人类渗入所致。研究中将这一未采样的古亚洲祖先成分命名为"Ypykuéra"(图皮语,意为"祖先")。
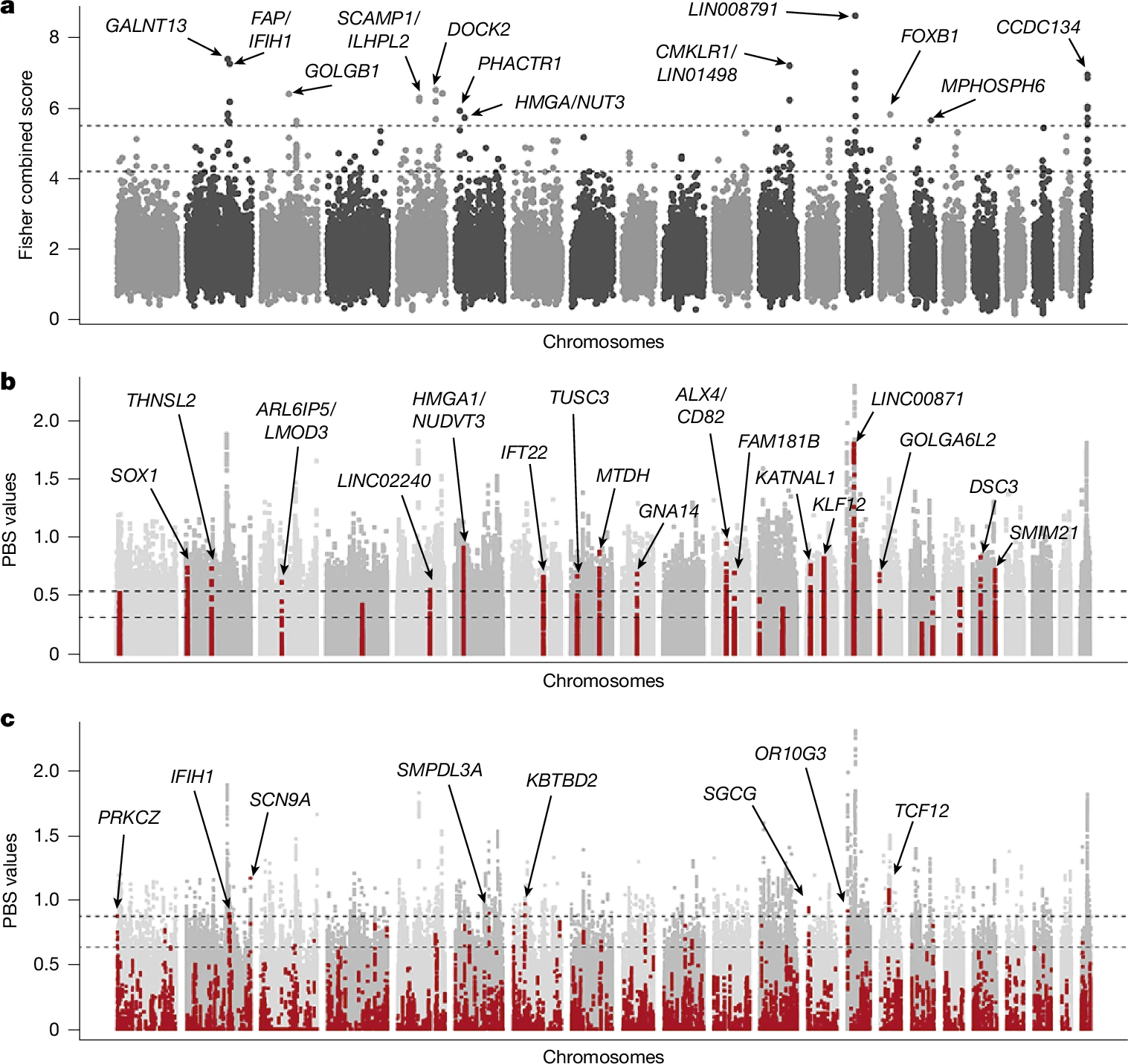
六、广泛的自然选择信号
通过 PBS + EHH 联合打分(Fisher combined score,滑动窗口 200 SNPs,步长 50 SNPs),在美洲原住民基因组中识别出 12 个候选选择区域,涉及功能包括:免疫应答(IFH1 ,病毒响应相关)、代谢与心血管表型(GALNT13 ,同时与胎儿生长和疟疾抵抗相关)、生育率(LINC00871,选择信号最强,与生育率相关 GWAS 位点重合)等。这些信号并非局限于某一生态区或特定人群,而是在整个美洲大陆范围内广泛分布。
另外,PBS 信号与澳洲亲缘性位点及古人类渗入位点的重叠分析显示,两类信号在基因组上的分布相当独立,进一步支持其独立的演化起源。
结论
本研究系统填补了美洲原住民高质量全基因组数据的空白,构建了迄今最全面的美洲原住民基因组多样性图谱。三次独立入迁模型取代了以往的单波扩散假说,揭示了南美洲人群构成远比预期更为复杂的历史。Ypykuéra 相关的澳洲血缘成分至少在一万年前就已存在,并可能在自然选择作用下得以长期维持。古人类适应性渗入同样在影响关键生物学功能方面留下了可检测的痕迹。殖民主义对原住民遗传多样性的破坏在 ROH 和 Ne 数据中有清晰的基因组印记。整体而言,这项研究对过于简化的美洲定居模型提出了有力挑战,也为未来基于原住民人群的生物医学研究奠定了重要的基因组基础。
参考文献
Castro e Silva, M.A., Nunes, K., Ribeiro, M.R. et al. The evolutionary history and unique genetic diversity of Indigenous Americans. Nature (2026). https://doi.org/10.1038/s41586-026-10406-w