作为细胞内无数生化反应的驱动力,蛋白质在细胞微观世界中扮演着建筑师和工程师的角色,不仅催化着生命活动,更是构筑、维系生物体形态与功能的基础构件。正是蛋白质之间的互动、协同作用,支撑起了生命的宏伟蓝图。
然而,蛋白质的结构复杂多变,传统的实验方法在解析蛋白质结构时既耗时又费力------蛋白质语言模型 (PLMs) 应运而生,利用深度学习技术,通过分析大量的蛋白质序列数据,学习蛋白质的生物化学规律和共进化模式,在蛋白质结构预测、适应性预测和蛋白质设计等领域取得了显著成就,极大地推动了蛋白质工程的发展。
尽管 PLMs 在残基尺度上取得了巨大成功,但在提供原子级信息方面的能力却受到了限制。针对于此,清华大学智能产业研究院副研究员周浩联合北京大学、南京大学和水木分子团队,提出了一种多尺度的蛋白质语言模型 ESM-AA (ESM All Atom), 通过设计残基展开、多尺度位置编码等训练机制,拓展出了处理原子尺度信息的能力。
ESM-AA 在靶点-配体结合等任务的性能显著提升,超越目前 SOTA 蛋白语言模型,如 ESM-2,也超越了目前的 SOTA 分子表示学习模型 Uni-Mol 等。相关研究已经以「ESM All-Atom: Multi-scale Protein Language Model for Unified Molecular Modeling」为题,发表于机器学习顶级会议 ICML 上。

论文地址:
https://icml.cc/virtual/2024/poster/35119
开源项目「awesome-ai4s」汇集了百余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
数据集:构建了蛋白质和分子数据的混合数据集
在预训练任务中,该研究使用了一个包含蛋白质和分子数据的组合数据集,其中包含原子坐标等结构信息。
对于蛋白质数据集,该研究使用了 AlphaFold DB ,其中包含了 800 万条高置信度的 AlphaFold2 预测的蛋白质序列和结构。
对于分子数据集,该研究使用了由 ETKDG 和 MMFF 分子力场生成的数据,包含 1,900 万个分子和 2.09 亿种构型。
在训练 ESM-AA 时,研究人员首先将一个蛋白质数据集 Dp 和一个分子数据集 Dm 混合在一起作为最终的数据集,即 D=Dp∪Dm 。对于来自 Dm 的分子而言,由于仅由原子组成,其代码转换序列 X̄ 就是所有原子 Ā 的有序集合,并且没有任何残基,即 R̄=∅。值得注意的是,因为在预训练中使用了分子数据,所以 ESM-AA 既可以接受蛋白质作为输入,也可以接受分子作为输入。
ESM-AA 模型构建:多尺度预训练与编码,实现统一分子建模
受多语言代码切换方法的启发,ESM-AA 在进行预测与蛋白质设计任务时,首先会随机解压缩部分残基,从而生成多尺度代码切换蛋白质序列,随后通过精心设计的多尺度位置编码对这些序列进行训练,并且已经在残基和原子尺度上证明了其有效性。
当处理蛋白质分子任务时,即涉及蛋白质和小分子的任务,ESM-AA 不需要任何额外模型辅助,可以充分发挥出预训练模型的能力。
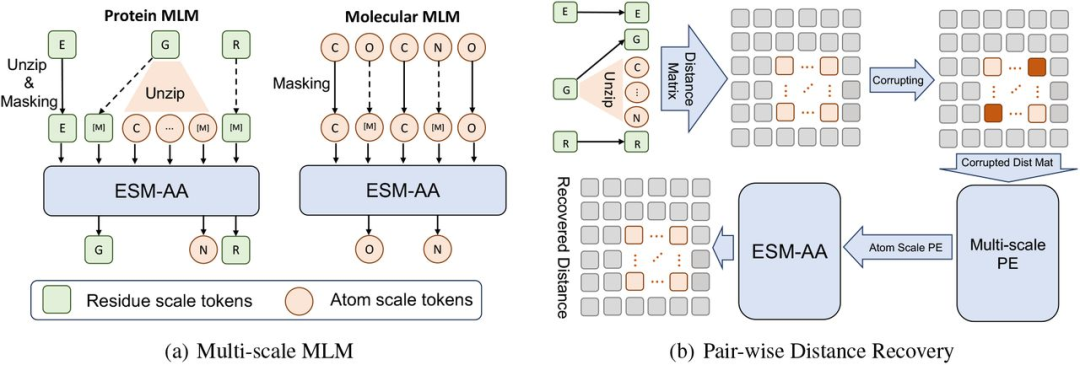
多尺度预训练框架
该研究的多尺度预训练框架由多尺度掩码语言建模 (masked language model, MLM) 和成对距离恢复 (pairwise distance recovery) 组成。
具体而言,在残基尺度上,一个蛋白质 X 可以被看作是一个由 L 个残基组成的序列,即 X = (r1,...,ri,...,rL)。每个残基 ri 都是由 N 个原子 A 组成 Ai={a1i,...,aNi}。为了构建代码切换蛋白序列 X̅,该研究通过随机选择一组残基,并将其对应的原子插入到 X 中,从而实现了一个解压缩的过程。在这一过程中,研究人员将被解压的原子按照顺序排列,最后在将原子集合 Ai 插入到 X 中(即解压残基 ri)后,即可得到一个代码切换序列 X̄。
随后,研究人员对代码切换序列 X̄ 进行掩码语言建模。
首先,随机遮挡 X̄ 中的一部分原子或残基,让模型使用周围上下文预测原始原子或残基。然后,研究人员使用对偶距离恢复 (PDR) 作为另一个预训练任务。即通过在坐标中添加噪声来破坏原子尺度的结构信息,并使用被破坏的原子间距离信息作为模型输入,要求模型恢复这些原子之间的准确欧几里得距离。
考虑到跨越不同残基的长距离结构信息与单个残基内部的原子尺度结构信息的语义差异,研究只计算残基内的 PDR,这也可以使 ESM-AA 学习到不同残基内的各种结构知识。
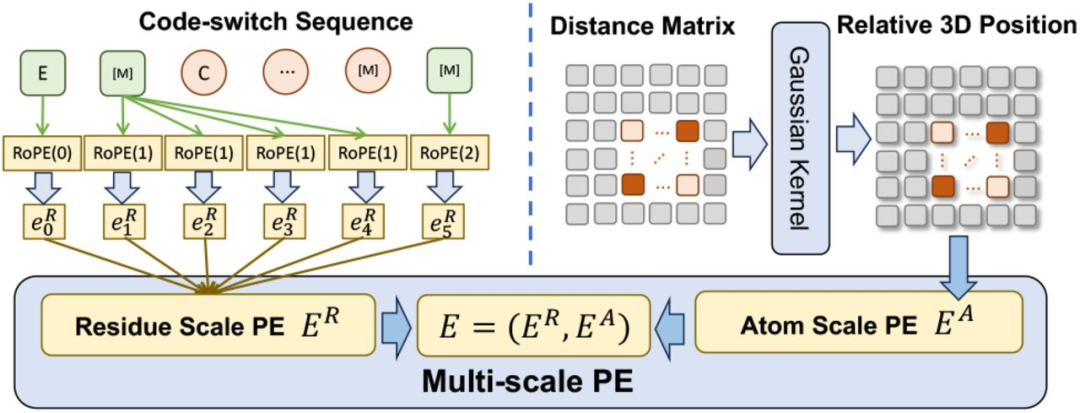
多尺度位置编码框架
在多尺度位置编码 (Multi-scale Position Encoding) 方面,研究人员设计了一个多尺度位置编码 E 来对代码切换序列中的位置关系进行编码。E 包含一个残基尺度的位置编码 ER 和一个原子尺度的位置编码 EA。
对于 ER, 研究人员扩展了现有的编码方法,使其能够编码从残基到原子的关系,同时在处理纯残基序列时保持与原始编码的一致性。对于 EA, 为了捕捉原子之间的关系,该研究直接使用空间距离矩阵 (spatial distance matrix) 对其三维位置进行编码。
值得一提的是,多尺度编码方法可以确保,预训练不会受到模糊位置关系的影响,从而使 ESM-AA 在两个尺度上都能有效地发挥作用。
在将多尺度 PE 集成到 Transformer 时,该研究首先用残差尺度位置编码 ER 替换了 Transformer 中的正弦编码,将原子尺度的位置编码 EA,视为自注意力层的偏置值 (bias term)。
研究结果:融合分子知识,优化蛋白质理解
为了验证多尺度统一预训练模型的有效性,该研究在各种涉及蛋白质和小分子的任务中评估了 ESM-AA 的表现。
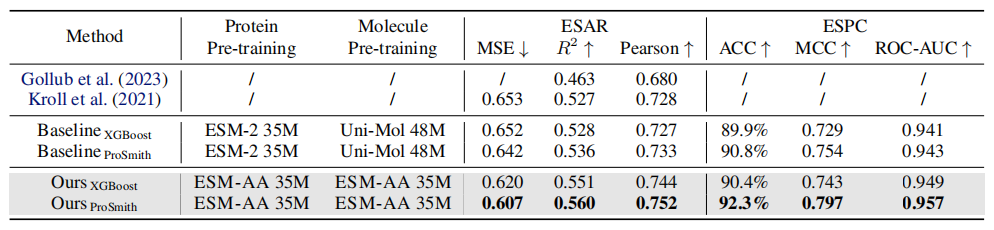
表1:在酶-底物亲和力回归任务 (ESAR) 和酶-底物对分类任务 (ESPC) 上的性能比较
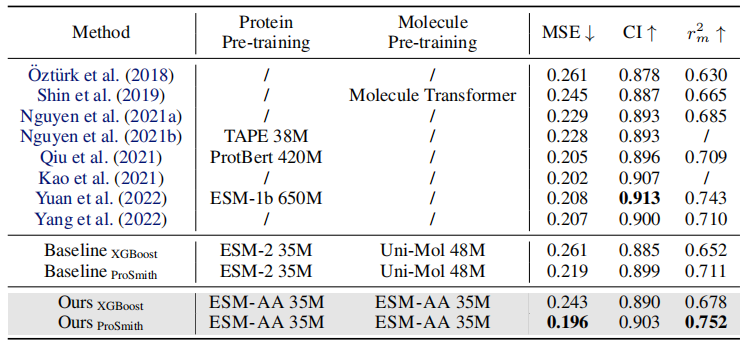
表2:药物-靶点亲和力回归任务的性能比较
如上表所示,在酶-底物亲和力回归任务、酶-底物对分类任务和药物-靶点亲和力回归任务的性能比较中,大多数指标上,ESM-AA 都优于其他模型并达到了最先进的结果。 此外,微调策略(如 ProSmith 和 XGBoost)建立在 ESM-AA 上时,性能始终优于结合两个独立的分子预训练模型与蛋白预训练模型的版本(如表 1 和表 2 最后四行所示)。
值得注意的是,ESM-AA 甚至可以打败使用了具有更大参数规模的预训练模型的方法(如表 2 中第 5 行、第 7 行与最后一行的对比)。
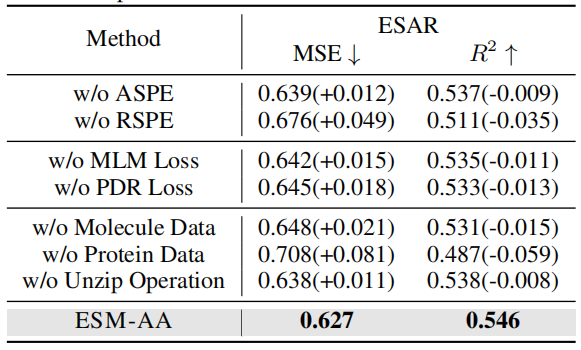
消融实验结果
为了验证多尺度位置编码的有效性,该研究在两种情况下进行了消融实验:一种是不使用原子尺度位置编码 (ASPE) 的情况;另一种是不使用遗传尺度位置编码 (RSPE) 的情况下进行。
在删除分子或蛋白质数据时,模型性能出现显著下降。有趣的是,删除蛋白质数据导致的性能下降比删除分子数据更明显。这表明,当模型没有经过蛋白质数据训练时,会迅速丢失与蛋白质相关的知识,从而导致整体性能明显下降。然而,即使没有分子数据,模型仍然可以通过解压缩操作获得原子级别的信息。
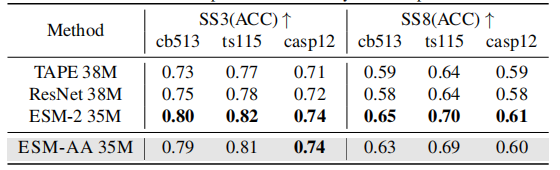
二级结构预测任务的性能比较
由于 ESM-AA 基于现有的 PLMs 开发,该研究希望确定其是否仍然保留了对蛋白质的全面理解,从而通过使用二级结构预测 (secondary structure prediction) 和无监督接触预测 (unsupervised contact prediction) 任务,测试蛋白质预训练模型在蛋白质结构理解方面的能力。
结果表明,虽然 ESM-AA 在此类研究中可能无法实现最佳性能,但其在二级结构预测和接触预测方面与 ESM-2 的表现相似。
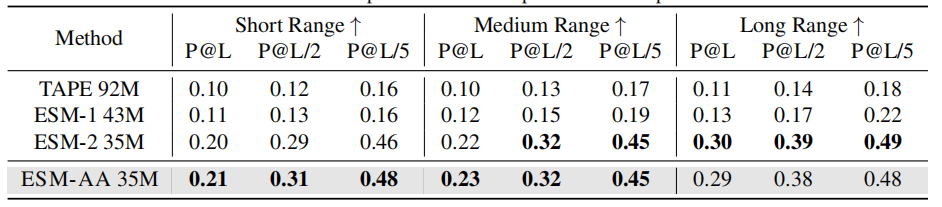
无监督接触预测任务的性能比较
在分子基准测试中,ESM-AA 在大多数任务中与 Uni-Mol 表现相当, 并在许多情况下优于几个特定于分子的模型,这表明其已成为一种处理分子任务的强大方法。

ESM-AA 和 ESM-2+Uni-Mol 学习表征的可视化
为了更直观地说明 ESM-AA 获得了更高质量的蛋白质和小分子表征,该研究在酶-底物对分类和药物靶点亲和力回归任务中,对 ESM-AA 和 ESM-2+Uni-Mol 提取的表征进行了可视化比较。结果显示,ESM-AA 模型能够创建包含蛋白质和分子数据的更具内聚性的语义表征,这使得 ESM-AA 优于两个单独的预训练模型。
蛋白质语言模型,大语言模型的下一段征程
大约从 1970 年代起,就有越来越多的科学家认为「二十一世纪是生物学的世纪。」去年 7 月,福布斯曾在一篇长文中畅想,LLM 令人们处于生物学领域新一轮变革的风口浪尖。生物学原来是一个可破译、可编程,在某些方面甚至是数字化的系统,LLM 凭借其对自然语言的惊人驾驭能力,为破解生物语言提供了潜在可能, 这也让蛋白质语言模型成为了这个时代最受关注的领域之一。
蛋白质语言模型代表了 AI 技术在生物学中的前沿应用。其通过学习蛋白质序列的模式和结构,能够预测蛋白质的功能和形态,对于新药开发、疾病治疗和基础生物学研究具有重大意义。
此前,蛋白质语言模型如 ESM-2 和 ESMFold 已经展现出与 AlphaFold 相媲美的准确性,并且具备更快的处理速度和对「孤儿蛋白质」更准确的预测能力。这不仅加速了蛋白质结构的预测,也为蛋白质工程提供了新的工具,使得研究人员能够设计出具有特定功能的全新蛋白质序列。
此外,蛋白质语言模型的发展受益于所谓的「缩放法则」,即模型的性能随着模型规模、数据集大小和计算量的增加而显著提高。 这意味着,随着模型参数的增加和训练数据的积累,蛋白质语言模型的能力将得到质的飞跃。
近两年,蛋白质语言模型在企业界也进入了快速发展时期。2023 年 7 月,百图生科与清华大学联合提出了一种名为 xTrimo Protein General Language Model (xTrimoPGLM) 的模型,参数量高达千亿 (100B),在多种蛋白质理解任务(15 项任务中的 13 项任务)中显著优于其他先进基线模型。在生成任务上,xTrimoPGLM 能够生成与自然蛋白质结构类似的新蛋白质序列。
2024 年 6 月,AI 蛋白质企业途深智合宣布,将其研发的国内首个自然语言蛋白质大模型 TourSynbio™ 面向所有科研人员和开发者开源。 该模型以对话的方式实现了对蛋白质文献的理解,包括蛋白质性质、功能预测和蛋白质设计等功能,在对比蛋白质评测数据集的测评指标上,超过 GPT4,成为行业第一。
此外,以 ESM-AA 为代表技术研究的突破,或许也意味着技术的发展即将度过「莱特兄弟时刻」,迎来飞跃。同时,蛋白质语言模型的应用也将不仅限于医疗和生物制药领域,还可能扩展到农业、工业、材料科学和环境修复等多个领域,推动这些领域的技术革新,为人类带来前所未有的变革。